Neutrophil Migration Assay:
Neutrophil granulocytes were isolated from heparinised peripheral blood of healthy donors using dextran sedimentation (3% dextran, added in 1:1 ratio) followed by Ficoll gradient separation. Residual red blood cells were lysed in hypotonic (0.2%) NaCl solution, followed by two washes in ice-cold PBS. The cells were thereafter suspended in Hanks’ balanced salt solution and 5% fatal bovine serum.
Cell migration was tested using 10 ng/ml recombinant human IL-8 (R&D Systems) as chemoattractant or empty buffer as negative control in Corning HTS Transwell 96-well permeable support plates with 3 micrometer pore size.
The number of migrated cells was studied following 60 minutes incubation at 37 °C with the help of flow cytometry. Cell numbers were calculated by using AccuCheck counting beads (Thermo Fisher Scientific).
Osteoclast (OC) cultures:
Monocytes were purified from peripheral blood (buffy coats) of healthy individuals. The buffy coats were obtained from the Karolinska University Hospital’s Blood Center. For the isolation of monocytes we used first Ficoll centrifugation to obtain PBMC, which was followed by magnetic separation (CD14 Microbeads, Miletnyi Biotech). Monocytes were cultured in 96-well flat bottom plates, 105 cells/200µl using DMEM (high glucose), complemented with 10% FBS, glutamine, penicillin-streptomycin (all from Sigma-Aldrich) and 25ng/ml recombinant human M-CSF (Peprotech).
On day 3, 50% of the medium was replaced, new medium with 20ng/ml M-CSF and 10ng/ml RANK-L (R&D Systems) was added (Note that the cytokines were 2X diluted on the plates since only 50% of the medium was replaced). Each sample was represented in triplicate wells. After every 3-day period, 50% of the medium was removed and new medium was added in addition to cytokines. The culture was stopped 6-9 days after the addition of RANK-L, as soon as osteoclasts become clearly visible using light microscope.
OCs were counted in light microscope as TRAP-positive cells with at least 3 nuclei. The staining reagent Acid Phosphatase, Leukocyte (TRAP) Kit (#387A) from Sigma-Aldrich was used for staining, with the following modifications: The assay was performed on 96 well plates, fixing solution was applied in 50µl/well, staining solution was applied in 60µl/well. Tartrate-negative stainings were not performed. Incubations were performed in incubator, at 37°C.
CXCR1 β-arrestin2 Assay:
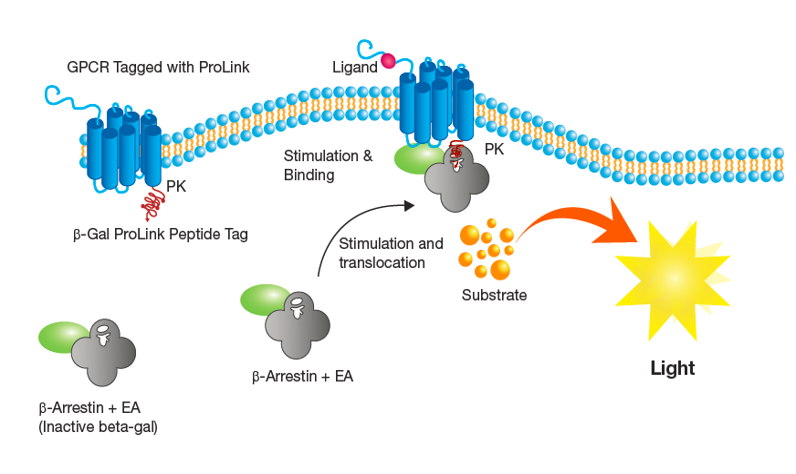
One vial of PathHunter® U2OS CXCR1 β-arrestin2 assay-ready cells (P/N 93-0226E3) was thawed in pre-warmed AssayComplete Cell Plating 0 Reagent (CP0; 93-0563R0) and cells (10K / well) were seeded in 100 µl / well into a 96-well clear bottom white-walled plate and allowed to recover for 48 hours in a humidified incubator at 37°C and 5% CO2.
Samples were prepared as 22X stocks in 11-pt dose response format, with 1:3 serial dilutions (highest concentration= 10µg/mL final) diluted in PBS + 0.1% BSA. Each dose was prepared in duplicate.
An EC-80 stock (840ng/mL final concentration) of IL-8 was prepared as a 22X stock in PBS + 0.1% BSA.
IL-8 stock was mixed in a 1:1 ratio with each antibody dilution and incubated at 37ºC for 15 minutes.
10 µl of each sample dilution / IL-8 mixture was added to assay plate containing cells and incubated for 1.5 hours in a humidified incubator at 37°C and 5% CO2.
To detect signal, 50 µL of PathHunter Detection Reagent (prepared per manufacturer’s instructions) was added to each well of the assay plate, prior to incubation of the plate at room temperature for 60 minutes in the dark.
Signal from each sample was acquired on a Perkin Elmer Envision plate reader with an integration time of 0.1 second/well. Raw RLU data were plotted using GraphPad Prism software. Data presented in graph format represent the average (+/- standard deviation) of duplicate wells per sample dose.



