This probe is available from Sigma, Tocris and Cayman Chemical.
| Probe |
 |
NI-57 |
A collaboration between the SGC and University College London (UCL) has resulted in the discovery of NI-57 as a chemical probe for the bromodomains of the BRPF (BRomodomain and PHD Finger) family of proteins (BRPF1/2/3).
BRPF1, BRPF2 (BRD1) and BRPF3 are scaffolding proteins, assembling HAT complexes of the MOZ/MORF family (MOZ, Ybf2/Sas3, Sas2, and Tip60) (1). These MYST complexes have a tetrameric core containing BRPF, the tumour suppressor ING and Eaf6/EPC (enhancer of polycomb)-related scaffold subunits. MYST complexes play crucial roles in DNA repair, recombination, and replication as well as in transcription activation (2,3). MOZ is frequently translocated in AML (acute myeloid leukemia) and is required for HSC proliferation (4).
Two BRPF1 isoforms (isoform A and B) can be generated by alternative splicing. In contrast to BRPF1B, the isoform A harbours a residue insertion in the ZA-loop that prevents binding to acetylated histone peptides (5).
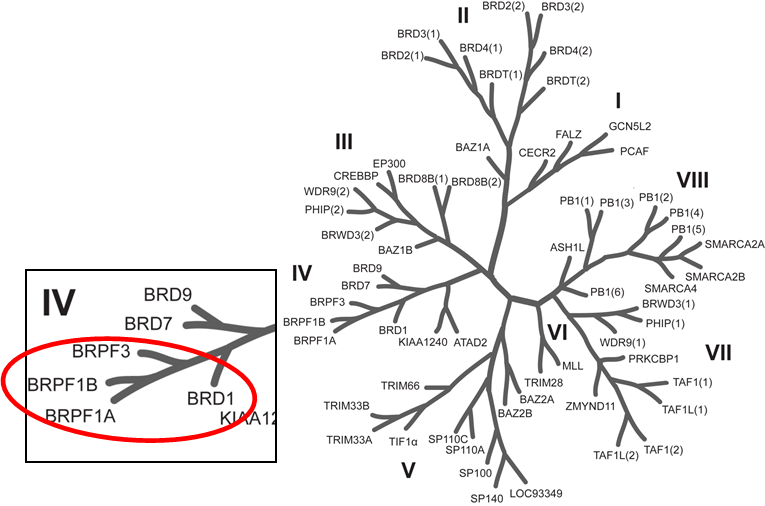
Phylogenetic tree of bromodomains and detailed view at the BRPF family.
NI-57 is a potent inhibitor of the bromodomain of the BRPFs and binds to BRPF1B with a KD of 31 nM (ITC), to BRPF2 with a KD of 108 nM (ITC) and to BRPF3 KD of 408 nM (ITC). NI-57 is very selective against other non-Class IV bromodomains, including the BETs, as measured by both biophysical and biochemical methods. The closest off-target effects are against BRD9 (32-fold selective). NI-57 shows accelerated FRAP recovery at 1 µM in the BRPF2 FRAP assay preventing binding of full-length BRPF2 to chromatin.
NI-57 is a chemical probe with potent binding affinity for BRPF1 (KD of 31 nM), BRPF2 (KD of 108 nM) and to BRPF3 (KD of 408 nM) with good selectivity over BRD9 (KD of 1 μM) and BRD4 (KD of 3.9 μM) as determined by isothermal titration calorimetry (ITC). Alpha screen biochemical assay confirmed NI-57 as a potent inhibitor of BRPF1 (IC50 of 114 nM). No off targets were identified for NI-57 outside the BRD family.
| Bromodomain | Kd/nM (ITC) | IC50/nM (Alpha Screen) | TM Shift °C |
| BRPF1B | 31 | 114 | 10.7±0.3 |
| BRPF2 (BRD1) | 110 | 619 | 5.6±0.1 |
| BRPF3 | 410 | 1010 | 5.3±0.3 |
| BRD9 | 1000 | 4400 | 3.1±0.7 |
| BRD4(1) | 3900 | >10,000 | 0.8±0.6 |
NI-57 showed significant selectivity for BRPF1/2/3 binding by DSF (differential scanning fluorimetry). NI-57 is very selective against other non-Class IV bromodomains, including the BETs, as measured by both biophysical and biochemical methods. The closest off-target effects were determined against BRD9 (32-fold selective). NI-57 shows minimal off-target pharmacology against a panel of GPCRs, ion channels, and enzymes and is functionally active in cellular assays.
We recommend to use NI-57 at concentrations of up to 1 µM and use the BRPF inhibitor OF-1 in parallel to confirm the result.
NI-57 increases thermal stability in the CETSA of full length BRPF1B but not of the BRPF1A isoform.
NI-57 induces dose-dependent displacement of BRPF1B but not of the BRPF1A isoform from histone H3.3 in NanoBRETTM assay with estimated IC50 values of 0.07 μM.
In a FRAP assay BRD1 as well as BRPF1B show reduced recovery time for the displacement of full-length from chromatin in the presence of NI-57.
NI-57 inhibits RANKL/MCSF induced differentiation of primary human monocytes into osteoclasts.
| Probe |
|
NI-57 |
| Physical and chemical properties | |
|---|---|
| Molecular weight | 383.42 |
| Molecular formula | C19H17N3O4S |
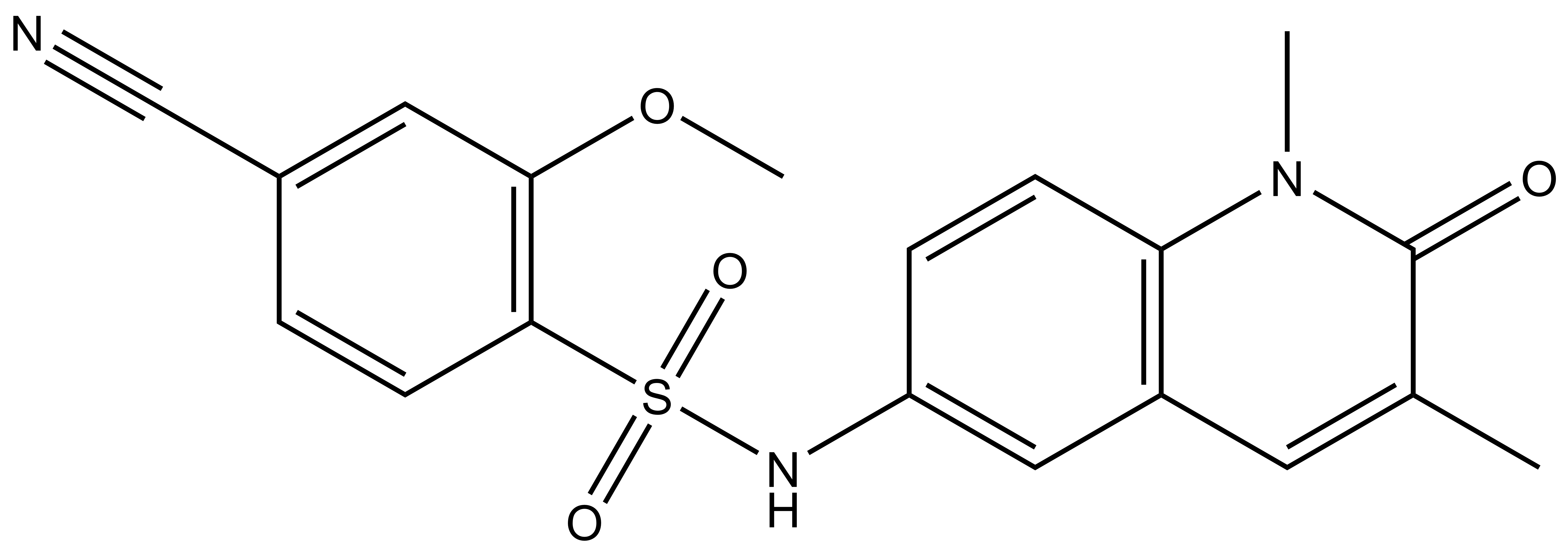
| IUPAC name | 4-cyano-N-(1,3-dimethyl-2-oxo-1,2-dihydroquinolin-6-yl)-2-methoxybenzenesulfonamide |
| logP | 2.05 |
| PSA | 99.5 |
| No. of chiral centres | 0 |
| No. of rotatable bonds | 5 |
| No. of hydrogen bond acceptors | 5 |
| No. of hydrogen bond donors | 1 |
| Storage | Stable as solid in the dark at -20°C. NB making aliquots rather than freeze-thawing is recommended |
| Dissolution | Soluble in DMSO |
Thermal Shift Assay

Selectivity profile of NI-57 using temperature shift assay. Temperature shifts ΔTm are indicated by red circles with increasing radii for higher Tm as described in the legend.
CEREP Screen
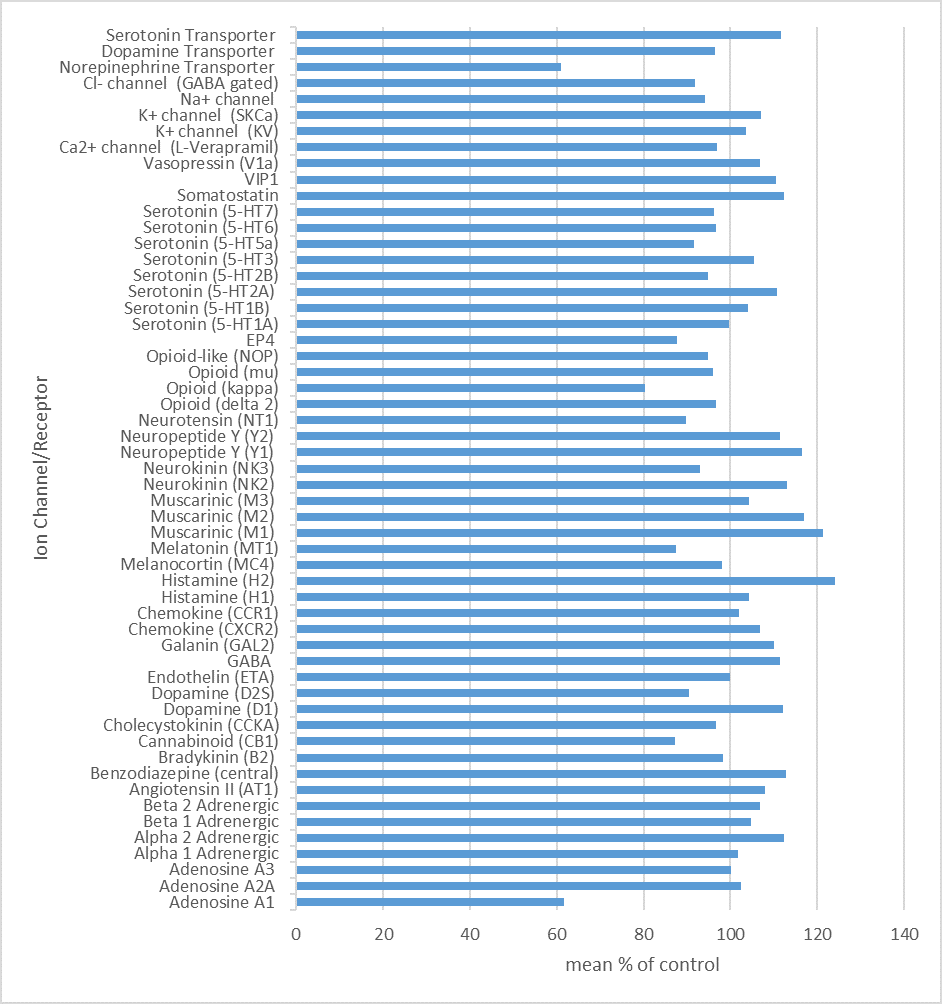
NI-57 (10 µm) was screened against a panel of 55 ligand receptors, ion channels and transports using an established and widely utilized commercial assay platform (ExpresSProfile; CEREP, Paris, FRANCE).
CESTA
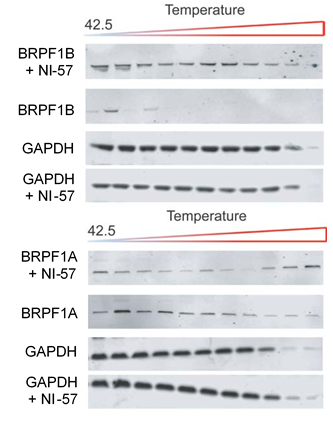
Thermal-stability of BRPF1A/B determined by CESTA upon application of NI-57.
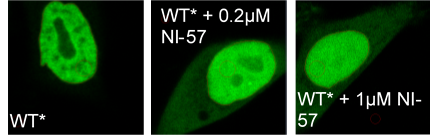
Re-location of BRPF1B to nucleolus and some ‘leak- out’ to cytoplasm in evidence upon compound treatment.
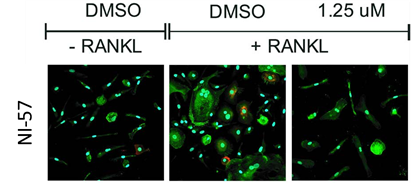
NI-57 attenuates osteoclastogenesis as seen when primary human monocytes (isolated from human cone blood via CD14+bead isolation) were cultured with MCSF (6 days) and the cells then treated with RANKL-/+ followed by NI-57 for at least 14 days. IF was performed with VNR (red), F-Actin (green) and DAPI to visualize the possible bone-resorbing cells
Work on this probe has been published in 'Design of a chemical Probe for the bromodomain and plant homeodomain finger-containing (BrPF) family of proteins' and 'Selective targeting of Bromodomain-PHD fingers family (BRPF) bromodomains impairs osteoclast differentiation'.
Isothermal Titration Calorimetry (ITC)
All calorimetric experiments were performed on a VP-ITC micro-calorimeter (MicroCalTM, LLC Northampton, MA). Protein solutions were buffer exchanged by gel filtration or dialysis into buffer (20 mM Hepes pH 7.5, 150 mM NaCl, and 0.5 mM tris (2-carboxyethyl) phosphine (TCEP). All measurements were carried out at 288.15 K. All injections were performed using an initial injection of 2 µL followed by injections of 8 µL. The data were analysed with the MicroCal ORIGIN software package employing a single binding site model. The first data point was excluded from the analysis.
Temperature shift assay
Thermal melting experiments were carried out using a Stratagene Mx3005p Real Time PCR machine (Agilent Technologies). OF-1 was added at a final concentration of 10 µM. SYPRO Orange (Molecular Probes) was added as a fluorescence probe at a dilution of 1:1000 as described (6).
AlphaScreen Assay
Assays were performed as described previously with minor modifications (7). All reagents were diluted in 25 mM HEPES, 100 mM NaCl, 0.1 % BSA, pH 7.4 supplemented with 0.05 % CHAPS and allowed to equilibrate to room temperature prior to addition to plates. An 11-point 1:2.0 serial dilutions of the ligands was prepared on lowvolume 384-well plates (ProxiPlateTM-384 Plus, PerkinElmer, USA), using LabCyte Eco liquid handler. Plates filled with 5 µL of the assay buffer followed by 7 µL of biotinylated peptide [H-YSGRGKacGGKacGLGKacGGAKacRHRK(Biotin)-OH for BRD1, BRD4, BRPF1B and BRPF3 or YQTARKSTGGK(ac)APRKQLATKAK(biotin)-OH for TIF1α] and Histagged protein to achieve final assay concentrations of 25-100 nM depending on the dose-response curve for each individual protein. Plates were sealed and incubated for a further 30 minutes, before the addition of 8 µM of the mixture of streptavidin-coated donor beads (12.5 µg/mL) and nickel chelate acceptor beads (12.5 µg/mL) under low light conditions. Plates were foil-sealed to protect from light, incubated at room temperature for 60 minutes and read on a PHERAstar FS plate reader (BMG Labtech, Germany) using an AlphaScreen 680 excitation/570 emission filter set. IC50 values were calculated in Prism 5 (GraphPad Software, USA) after normalization against corresponding DMSO controls.
Fluorescence Recovery After Photobleaching (FRAP) Assay
FRAP studies were performed using U20S cells expressing full-length BRPF2 (BRD1). Six hours after transfection 2.5 µM SAHA (to increase global histone acetylation) was added and cells were treated with 1 µM or 5 µM of OF-1 1 hour before imaging and half recovery times from the fluorescence signal of the bleached U2OS nuclei were plotted.
NanoBRET
U2OS cells were co-transfected with Histone H3.3-HaloTag and NanoLuc-BRPF1. Twenty hours post-transfection cells were collected, washed with PBS, and exchanged into media containing phenol red-free DMEM and 4% FBS in the absence (control sample) or the presence (experimental sample) of 100 nM NanoBRET 618 fluorescent ligand (Promega). Cells were then treated with an increasing dose of OF-1. Five minutes prior to reading, NanoBRET furimazine substrate (Promega) was added to both control and experimental samples and plates were read on a CLARIOstar (BMG) equipped with 450/80 nm bandpass and 610 nm longpass filters with a 0.5 sec reading setting. A corrected BRET ratio was calculated and is defined as the ratio of the emission at 610 nm/450 nm for experimental samples (i.e. those treated with NanoBRET fluorescent ligand) subtracted by and the emission at 610 nm/450 nm for control samples (not treated with NanoBRET fluorescent ligand). BRET ratios are expressed as milliBRET units (mBU), where 1 mBU corresponds to the corrected BRET ratio multiplied by 1000. Relative IC50 values were estimated by non-linear regression analysis of (log) concentration of each inhibitor versus milliBRET ratios (GraphPad Prism).
Human osteoclast differentiation
Primary human peripheral blood mononuclear cells (PBMCs) were collected from a Histopaque generated buffy coat after gradient centrifugation at 20 min and 500g, brakes off. The CD14+ monocyte fraction was obtained by on-column CD14+-MACS bead isolation, washed twice with MACS buffer, and seeded at a density of 50 000 c/mL in αMEM/10%FCS supplemented with 25 ng/mL MCSF. After 6 days at 37 °C, 5% CO2 treatment with NI-57 with and without 50 ng/mL RANKL was started. Media were changed with fresh compound every 3−4 days. After 14−21 days, cells were fixed and stained. IF was performed with VNR (red), F-Actin (green) and DAPI to visualize the possible bone-resorbing cells