The probe DDR-TRK-1 is available from Cayman Chemical and Sigma.
| Probe | Negative control | |
 |
|  |
DDR-TRK-1 |
| DDR-TRK-1N |
The discoidin domain receptors (DDRs), DDR1 and DDR2, are unique among the receptor tyrosine kinases (RTKs) in being activated by interaction with the extracellular matrix via binding to triple-helical collagen by the receptor extracellular domains.1 DDR1 and DDR2 form constitutive dimers making them unusual among RTKs, which typically dimerize only upon activation.2 DDRs regulate extracellular matrix remodeling, as well as cell adhesion, proliferation and migration.3 DDR kinases are linked to the progression of various human diseases, including fibrotic disorders, atherosclerosis and cancer.3-5 Significantly, they are identified as indicators of poor prognosis in ovarian, breast and lung cancer.6 DDR1 overexpression is associated with increased cell survival and invasion in hepatocellular carcinomas, pituitary adenoma and prostate cancer,7 whereas DDR2 is mutated in squamous cell lung cancers8 and contributes to breast cancer metastasis.9 The promise of DDR kinases as a therapeutic target has been demonstrated by DDR1 knockdown that has been shown to reduce metastatic activity in lung cancer models,10 slow the development of atherosclerosis,5 and impede the development of fibrotic disorders.11
The TRK kinases are represented by three members, TRKA, TRKB, and TRKC, which are selectively expressed in neuronal tissue. Receptor signaling is initiated by binding of the neurotrophic factors NGF, BDNF, and NTR, respectively. Subsequent signaling is via the RAS, PLCγ and PI3γ pathways. The TRKs play a vital role in CNS development and survival. Gene fusions, protein overexpression, and single nucleotide alterations, have been implicated in the pathogenesis of specific cancer types including glioblastoma, papillary thyroid carcinoma, and secretory breast carcinomas, but are rare in most other cancers.12
DDR-TRK-1 is a chemical probe for the DDR and TRK kinases with good in vitro and in vivo potency.
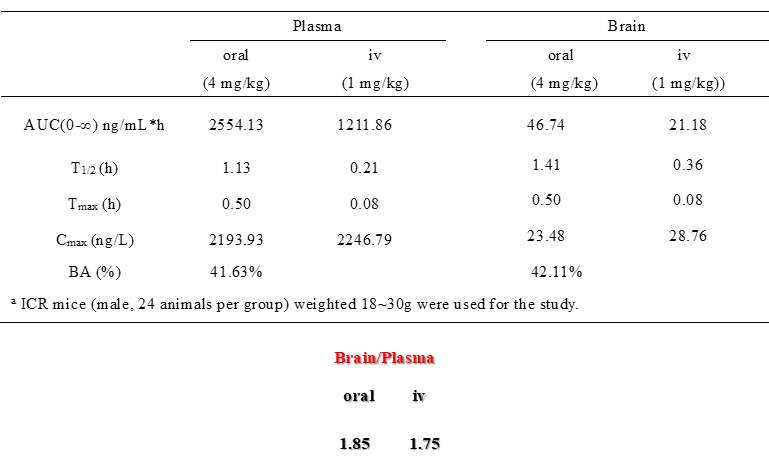
DDR-TRK-1 inhibits colony formation and migration of Panc-1 pancreatic cancer cells. In cellular and mouse models of lung fibrosis, DDR-TRK-1 inhibits signaling, expression of fibrotic markers and fibrotic features such as hydroxproline expression. With restricted CNS exposure and therefore no TRK inhibition, the in vivo effects of DDR-TRK-1 can be attributed to DDR1-2 inhibition. DDR-TRK-1N is the negative control compound with very minimal differences in compound structure, and should be used in parallel to DDR-TRK-1. The probe can be complemented by the use of BAY-826, which inhibits the kinases TIE1, TIE2, DDR1 and DDR2, but lacks TRK activity to better understand the target involved in the phenotypic effect.
Work on this probe has been published in J.Med.Chem. 2016 59(12), p 5911-5916, 'Structure-Based Design of Tetrahydroisoquinoline-7-carboxamides as Selective Discoidin Domain Receptor 1 (DDR1) inhibitors'.
DDR-TRK-1 is a chemical probe for the DDR and TRK kinases (IC50 3-43 nM) with corresponding good cellular potency in NanoBRETTM target engagement assays (IC50 104-448 nM). DDR-TRK-1 was shown to be selective in an in vitro kinase panel followed by cellular NanoBRETTM assays.
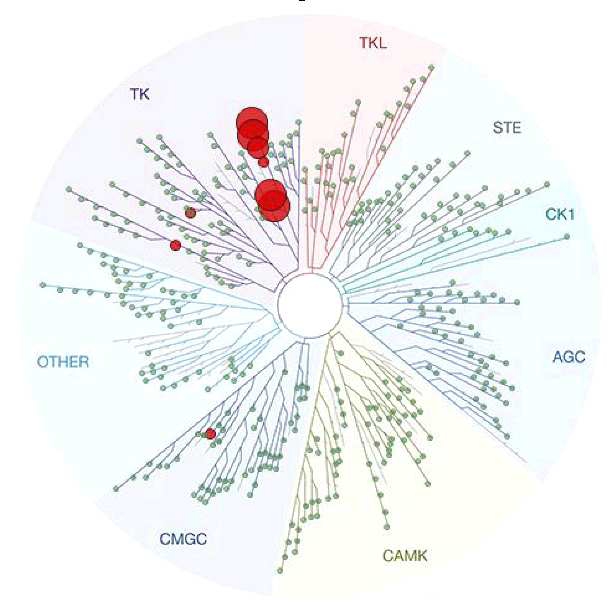
DDR-TRK-1 is selective in KINOMEscan® at 1μM. The closest off target is CDK11 (370 nM), which was however shown to be only poorly inhibited in cells (~5 µM in NanoBRETTM). The negative control DDR-TRK-1N is entirely clean in KINOMEscan at 1μM.
We recommend that DDR-TRK-1 be used at 5μM concentration in cells. The negative control DDR-TRK-1N should be used at 5μM concentration in cells – it is toxic in HeLa cells above 10μM.
We also recommend the use of selective TRK inhibitors or the TIE-DDR BAY-826 probe in parallel to dissect the biology of DDR1/2 versus TRKA/B/C.
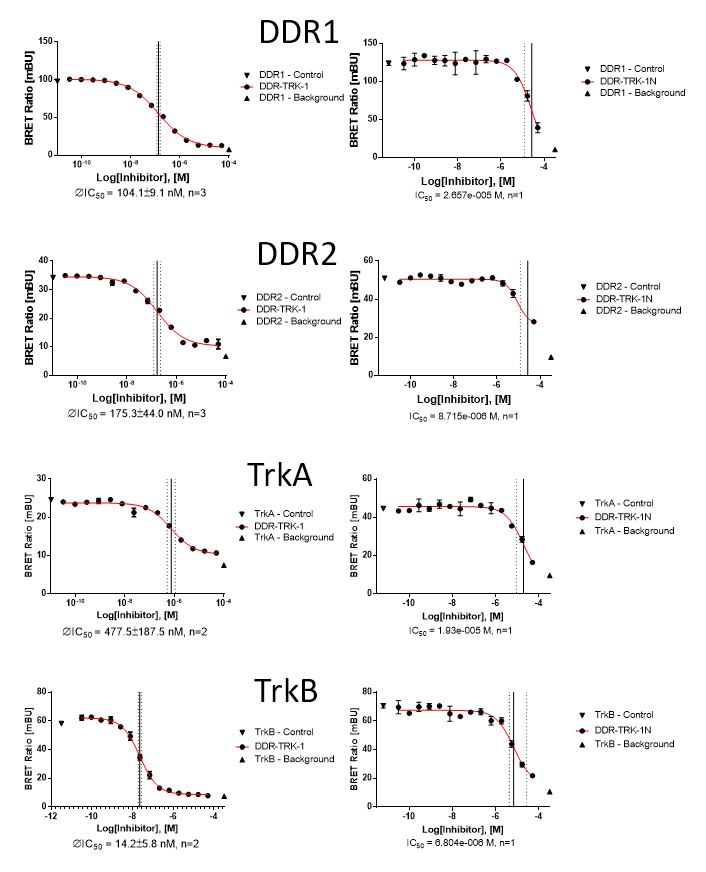
In NanoBRETTM assays, DDR-TRK-1 shows a potency of 104nM against DDR1, 175nM against DDR2, 448nM against TRKA and 142nM against TRKB.
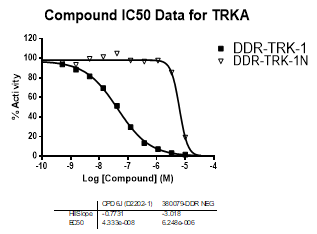
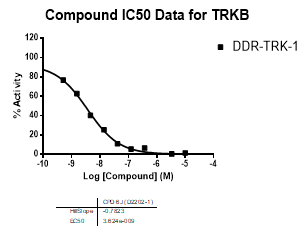
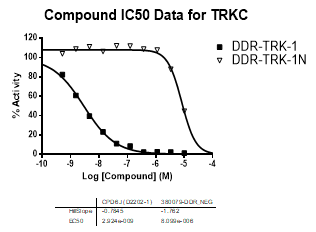
In an activity assay at RBC (10μM ATP), DDR-TRK-1 shows an IC50 value of 27nM against DDR1, 4.5nM against DDR2, 43nM against TRKA, 3.6nM against TRKB and 2.9nM against TRKC.
| Probe | Negative control | |
|
| |
DDR-TRK-1 |
| DDR-TRK-1N |
| Click here to download the DDR-TRK-1 SDF file. | Click here to download the DDR-TRK-1N SDF file. |
| Physical and chemical properties for DDR-TRK1 | |
| Molecular weight | 492.1885 |
| Molecular formula | C29 H31 F3 N4 O |
| IUPAC name | (3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoro-methyl)-phenylamino)-(5-methyl-3-(pyrimidin-5-yl)-3-aza-bicyclo[4.4.0]deca-1(6),7,9-trien-9-yl)-methanone |
| MollogP | 4.6 |
| PSA | 59.2 |
| No. of chiral centres | 1 |
| No. of rotatable bonds | 6 |
| No. of hydrogen bond acceptors | 5 |
| No. of hydrogen bond donors | 1 |
| Storage | -20 as DMSO stock |
| Dissolution | Soluble in DMSO at least up to 50mM |
| Physical and chemical properties for DDR-TRK-1N | |
| Molecular weight | 508.2450 |
| Molecular formula | C26 H23 F3 N6 O |
| IUPAC name | (3-((4-methyl-piperazin-1-yl)-methyl)-5-(trifluoro-methyl)-phenylamino)-(3-phenyl-3-aza-bicyclo[4.4.0]deca-1(6),7,9-trien-9-yl)-methanone |
| MollogP | 5.6 |
| PSA | 33.6531 |
| No. of chiral centres | 0 |
| No. of rotatable bonds | 7 |
| No. of hydrogen bond acceptors | 4 |
| No. of hydrogen bond donors | 1 |
| Storage | -20 as DMSO stock |
| Dissolution | Soluble in DMSO at least up to 50mM |
SMILES:
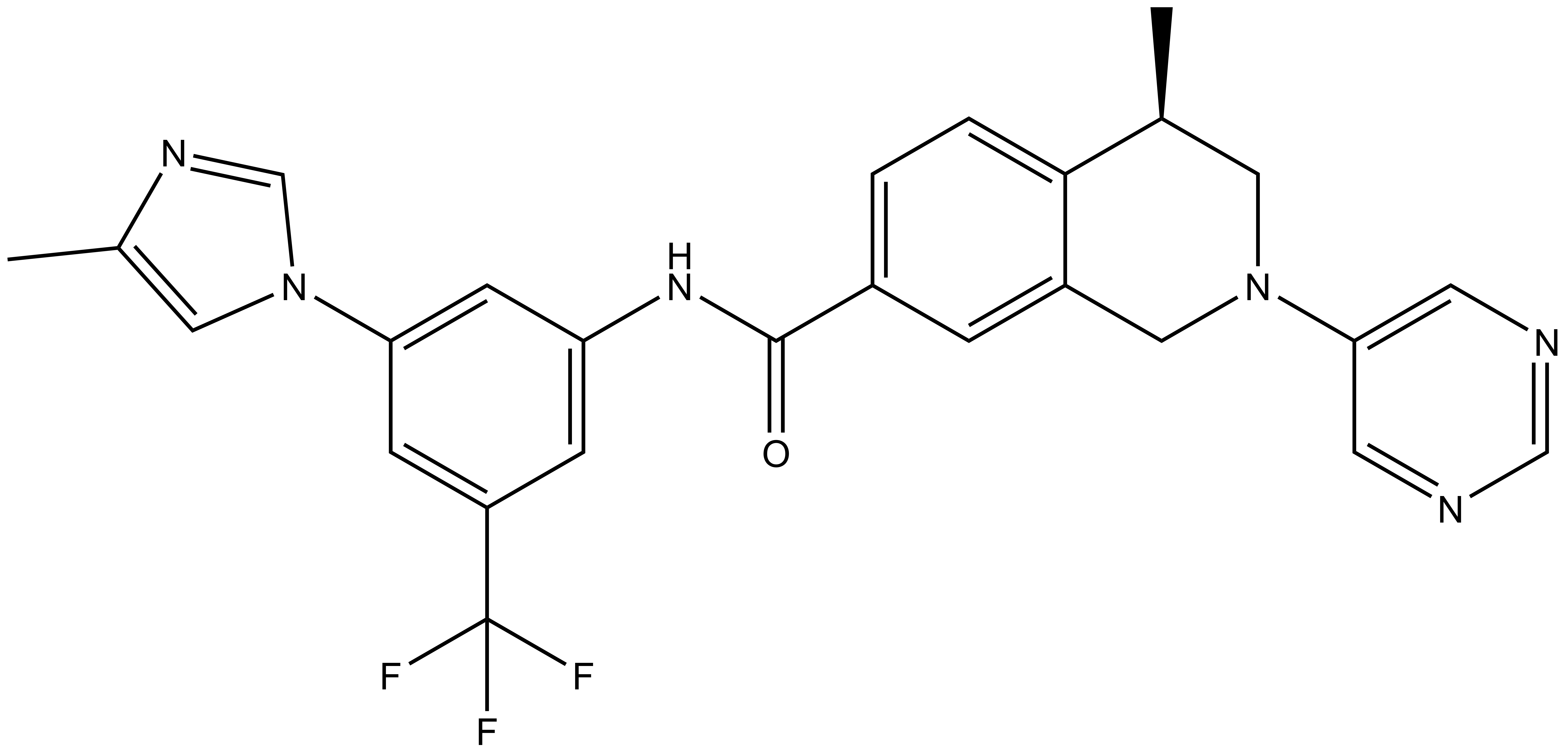
DDR-TRK-1: CC1=CN(C2=CC(NC(C3=CC=C4C(CN(C5=CN=CN=C5)C[C@@H]4C)=C3)=O)=CC(C(F)(F)F)=C2)C=N1
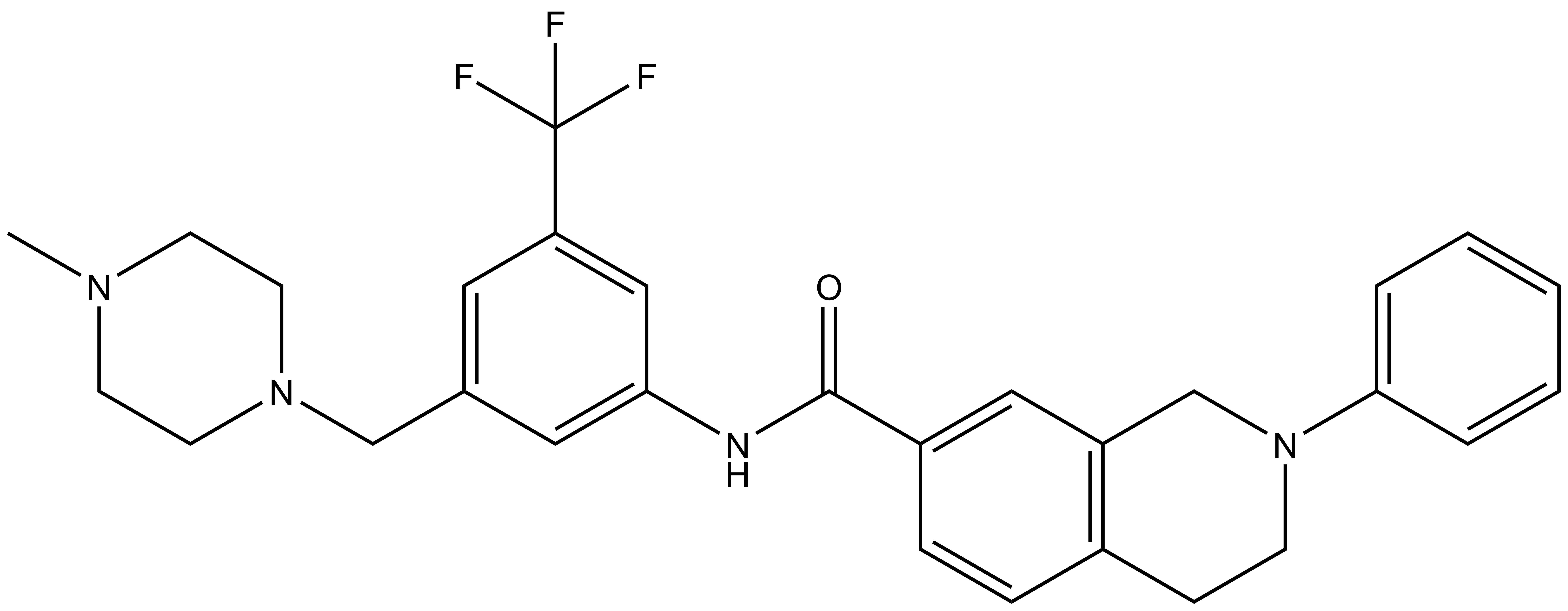
DDR-TRK-1N: CN1CCN(CC2=CC(C(F)(F)F)=CC(NC(C3=CC4=C(CCN(C5=CC=CC=C5)C4)C=C3)=O)=C2)CC1
InChI:
DDR-TRK-1: InChI=1S/C26H23F3N6O/c1-16-11-34(23-9-30-14-31-10-23)13-19-5-18(3-4-24(16)19)25(36)33-21-6-20(26(27,28)29)7-22(8-21)35-12-17(2)32-15-35/h3-10,12,14-16H,11,13H2,1-2H3,(H,33,36)/t16-/m0/s1
DDR-TRK-1N: InChI=1S/C29H31F3N4O/c1-34-11-13-35(14-12-34)19-21-15-25(29(30,31)32)18-26(16-21)33-28(37)23-8-7-22-9-10-36(20-24(22)17-23)27-5-3-2-4-6-27/h2-8,15-18H,9-14,19-20H2,1H3,(H,33,37)
InChIKey:
DDR-TRK-1: CMJJZRAAQMUAFH-INIZCTEOSA-N
DDR-TRK-1N: FWKRCZKMCJOFNG-UHFFFAOYSA-N

A KINOMEscan of DDR-TRK-1 at 1μM revealed very few off-targets:
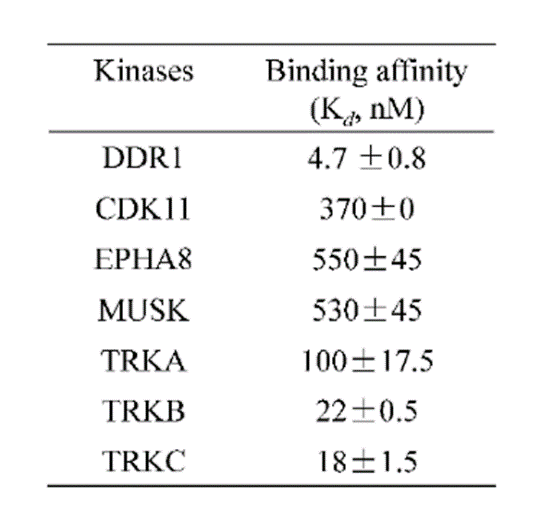
An activity assay at RBC (10μM ATP) revealed the following within the DDR/TRK family:
| Compound IC50 (M): | ||||
| Kinase: | DDR-TRK-1 | DDR-TRK-1N | Window probe/control | |
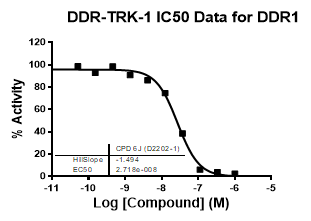
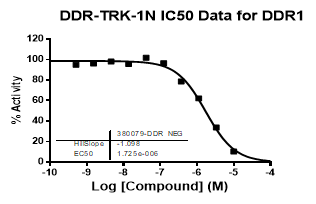
| DDR1 | 2.72E-08 | 1.73E-06 | 24 | |
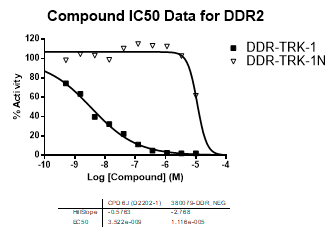
| DDR2 | 3.52E-09 | >1.00E-05 | NA | |
| TRKA | 4.33E-08 | 6.25E-06 | 144 | |
| TRKB | 3.62E-09 | NA | ||
| TRKC | 2.92E-09 | 8.10E-06 | >2000 | |
| Dose-response curves for the probe (and where collected, the negative control) are shown below: |
 |
 |
 |
 |
 |
 |
In a NanoBRETTM cellular target engagement assay DDR1-TRK-1 displayed dose dependant inhibition as follows:
| DDR-TRK-1 (M) | DDR-TRK-1N (M) | Window | |
| DDR1 | 1.04E-07 | 2.66E-05 | 256 |
| DDR2 | 1.75E-07 | 8.72E-06 | 50 |
| TRKA | 4.48E-07 | 1.93E-05 | 43 |
| TRKB | 1.42E-08 | 6.80E-06 | 478 |
NanoBRET assay
Dose-response experiments were conducted in 96 well format using HEK293T cells expressing NanoLuc fused to the C-terminus of full-length protein kinases for DDR2, TRKA and TRKB or isoform 2 for DDR1, using Promega tracer as indicated below.
| Kinase | Nluc location | full-length? | Tracer | [Tracer], nM (@Tracer) |
| DDR1 | C | Isoform 2 (Uniprot Q08345-2) | 4 | 85 |
| DDR2 | C | canonical (Uniprot Q16832-1) | 4 | 45 |
| TrkA | C | canonical (Uniprot P04629-1) | 5 | 50 |
| TrkB | C | canonical (Uniprot Q16620-1) | 5 | 30 |
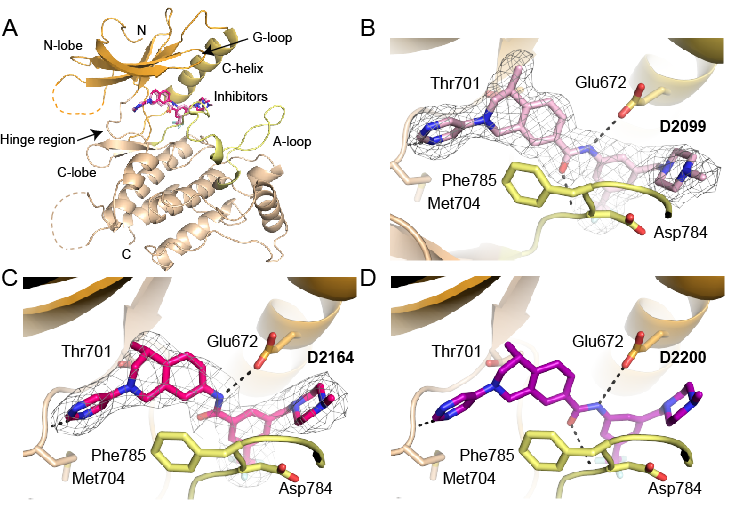
Structures of DDR1 complexes. A. Overall view of DDR1. Structural features are labelled for clarity. N-lobe is coloured orange, A loop is marked in yellow and C-lobe is coloured wheat. B. D2099 compound electron density (PDB 5FDP). C. D2164 compound electron density (PDB 5FDX). D. Molecular docking of DDR-TRK-1 compound.
Inhibition of DDR1, DDR2, TRKA, TRKB and TRKC kinase activity was measured using radiometric assay (Reaction Biology Corporation). Compounds were tested in 10-dose IC50 singlicate mode with a 3-fold serial dilution starting at 1 or 10 μM at [ATP] = 10 µM.
Kinome-wide profiling was performed at DiscoverX using the KINOMEscan assay.