This probe is available from Cayman Chemical and Tocris.
| Probe | Negative control | |
 |
|  |
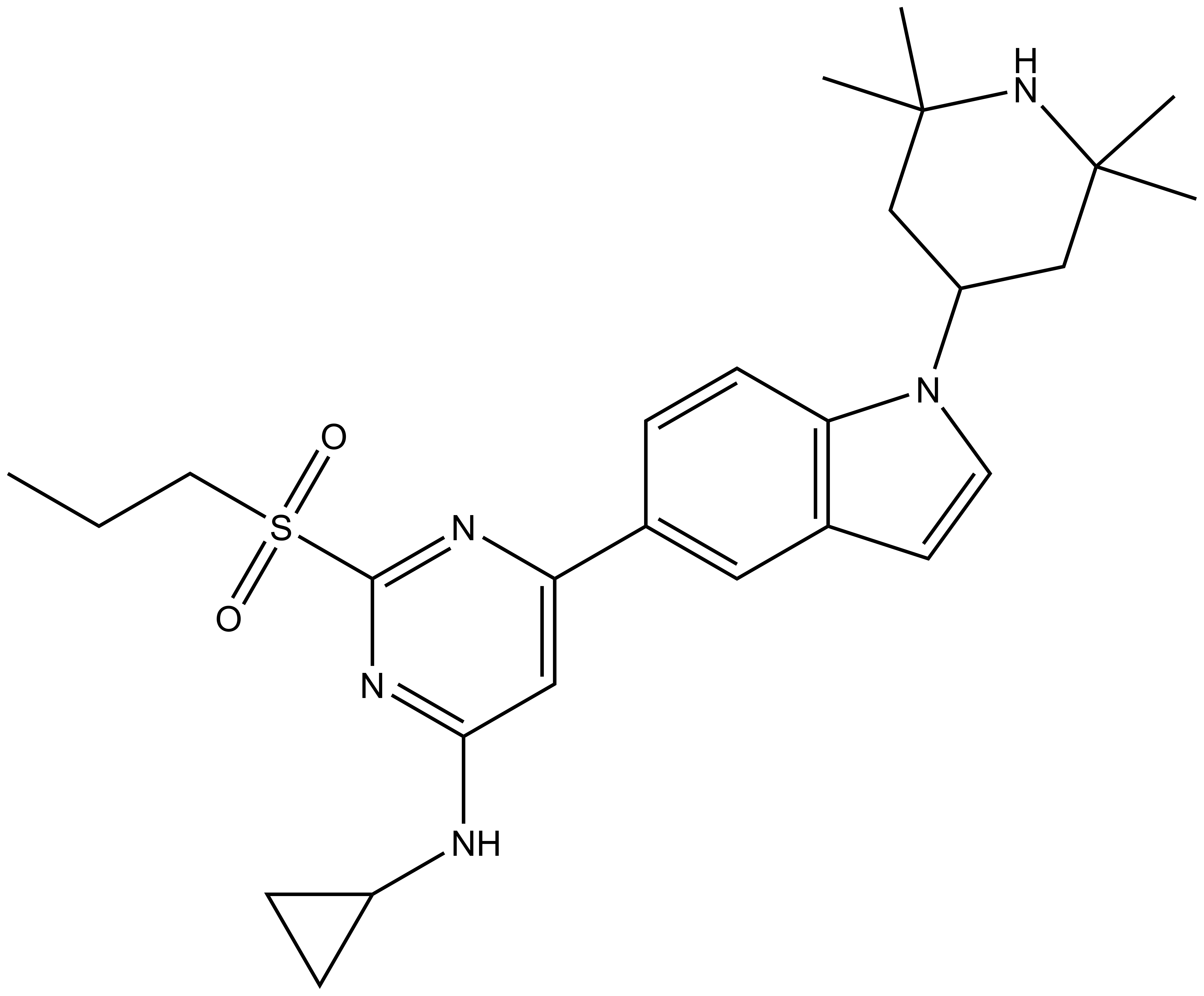
NVS-CECR2-1 |
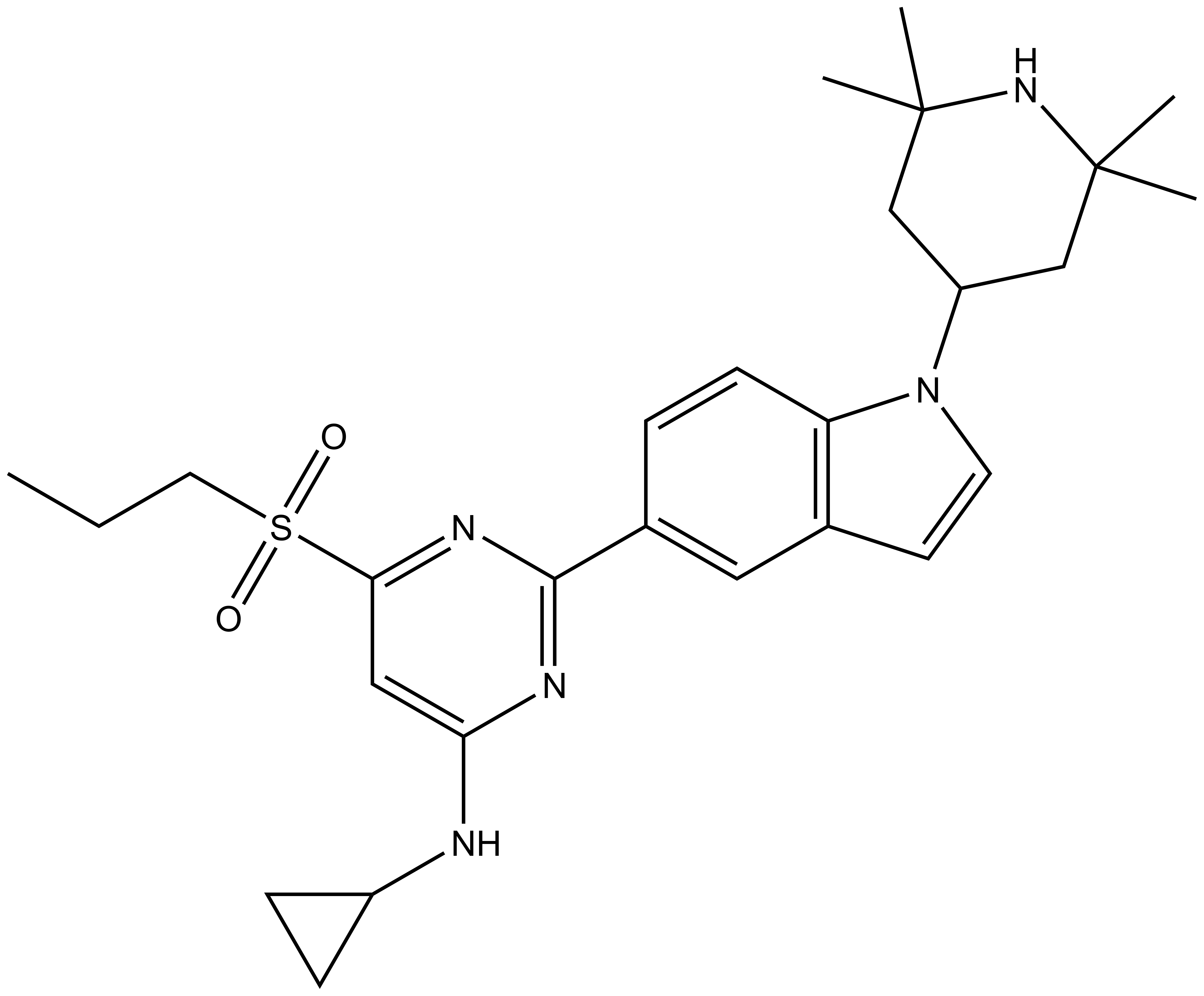
| NVS-CECR2-C |
NVS-CECR2-1 is a highly potent and selective CECR2 inhibitor that has been developed in collaboration with Novartis. CECR2 (cat eye syndrome chromosome region, candidate 2) gene previously was identified as being in the chromosome 22q11 region, duplicated in the human disorder cat eye syndrome (1). This syndrome is characterized by defects of the eye, heart, anus, kidney, skeleton, face and mental development; however the neural tube develops normally. CECR2 deletion in mice causes neural tube defects including severe exencephaly and perinatal death (2). CECR2 is predominantly expressed in the nervous system and involved in neurulation. Chromatin remodelling complexes play critical roles in development and CECR2 has been shown to be part of the CERF complex with SNF2L forming an ATP-dependent chromatin remodeller (2). CECR2 shows complex alternative splicing, but all variants contain DDT and bromodomain motifs. CECR2 has also been suggested to play a role in DNA damage response by inhibiting γ-H2AX (3).
NVS-CECR2-1 binds to CECR2 with high affinity (IC50 of 47 nM in alpha screen, KD = 80 nM in ITC), and demonstrates no cross reactivity in a BRD panel (48 targets). In the FRAP assay at 0.1 µM NVS-CECR2-1 shows robust activity in cells due to its slow off-rate, but no acute toxicity. No major activity is observed in kinase, protease and receptor panels.
NVS-CECR2-1 is poorly soluble but due to its high potency it may be safely used at low concentrations in cell biology applications. The structurally related NVS-CECR2-C is a suitable control compound that is inactive against CECR2.
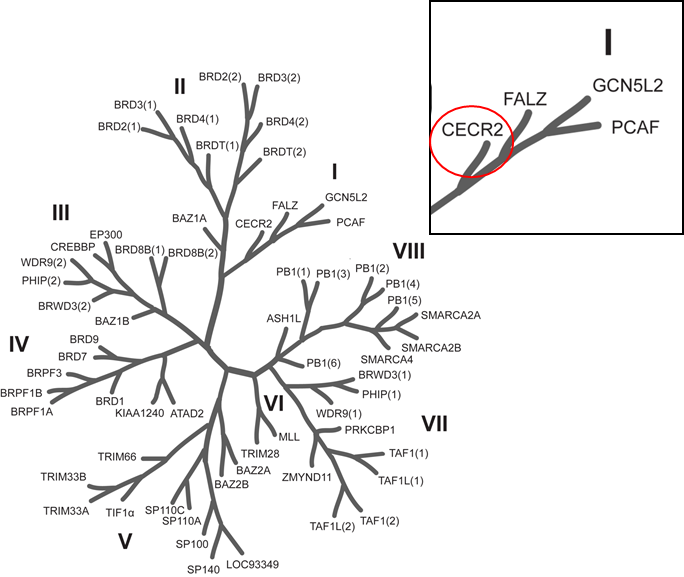
Phylogenetic tree of bromodomains and detailed view at CECR2.
| Bromodomain | Kd/nM (ITC) | IC50/nM (Alpha Screen) | TM Shift °C |
| CECR2 | 80 | 47 | 12.52 |
| BRD4 | NT | >37,000 | 1.73 |
| BRD7 | NT | 5500 | 1.21 |
| BRD9 | NT | 2300 | NT |
NVS-CECR2-1 is a highly potent inhibitor for CECR2 with a KD of 80 nM and shows very good potency in FRAP assay at 0.1 µM due to its slow off-rate. Alpha Screen confirmed NVS-CECR2-1 as a potent inhibitor of CECR2 with a IC50 of 47 nM.
NVS-CECR2-1 induced significant ΔTm shifts (12.52 °C). Only weak interactions were observed for BRD4 (1.73 °C) and BRD7 (1.21 °C), however alpha screen did not reveal strong interactions. NVS-CECR2-1 shows no cross reactivity among bromodomains and no significant inhibition of protein kinases, proteases and receptors.
Use at concentrations of up to 1 µM in cellular assays. The structurally related NVS-CECR2-C is a suitable control compound that is inactive against CECR2.
In a NanoBRETTM assay, CECR2 shows dose-dependent displacement from histone H3.3, with IC50 of 255 nM and the FRAP assay reveals robust inhibition of CECR2 at 0.1µM concentration of NVS-CECR2-1.
| Probe |
|
NVS-CECR2-1 |
| Physical and chemical properties | |
|---|---|
| Molecular weight | 495.68 |
| Molecular formula | C27H37N5O2S |
| IUPAC name | N-cyclopropyl-2-propylsulfonyl-6-[1-(2,2,6,6-tetramethylpiperidin-4-yl)indol-5-yl]pyrimidin-4-amine |
| clogP | 4.5 |
| PSA | 97.3 |
| No. of chiral centres | 0 |
| No. of rotatable bonds | 7 |
| No. of hydrogen bond acceptors | 6 |
| No. of hydrogen bond donors | 2 |
| Storage | stable as solid in the dark at -20°C. NB making aliquots rather than freeze-thawing is recommended |
| Dissolution | Soluble in DMSO |
Isothermal Titration Calorimetry
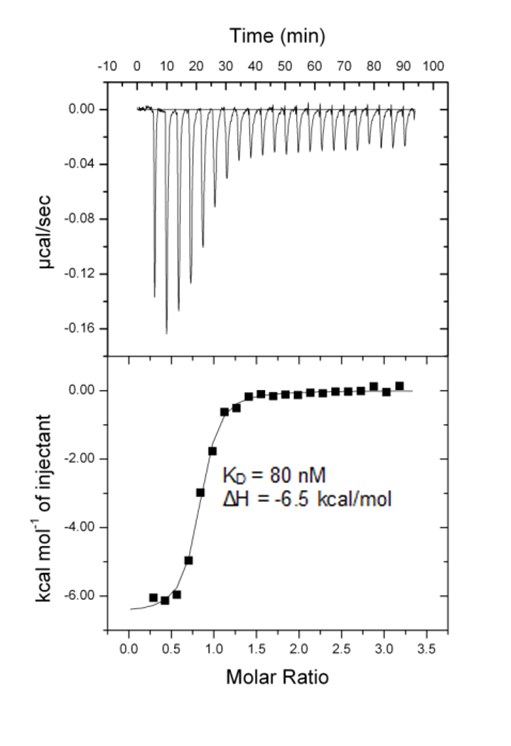
ITC measurements of the NVS-CECR2-1 with CECR2.
Temperature Shift Assay
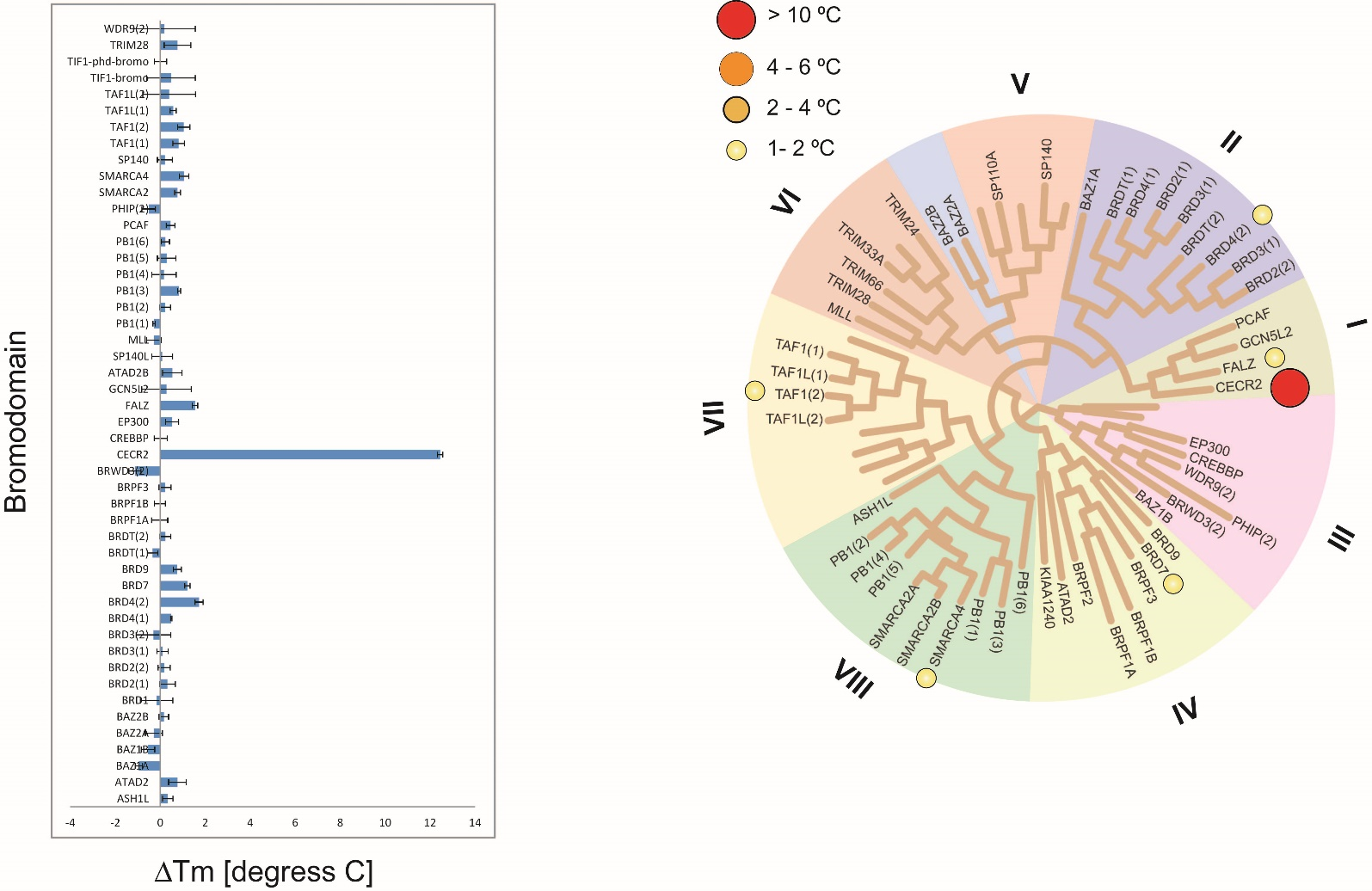
Selectivity screening of chemical probe NVS-CECR2-1 determined by temperature shift assay. The temperature shifts were mapped onto the phylogenetic tree using red circles corresponding to ΔTm as indicated in the figure.
NanoBRET
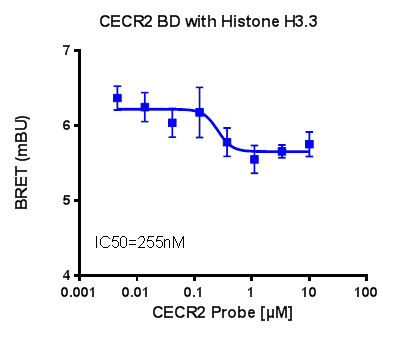
Dose-dependent displacement of CECR2 from histone H3.3 following treatment with NVS-CECR2-1.
Fluorescence Recovery After Photobleaching
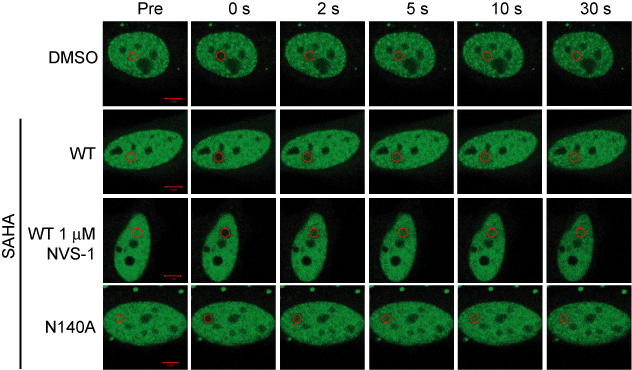
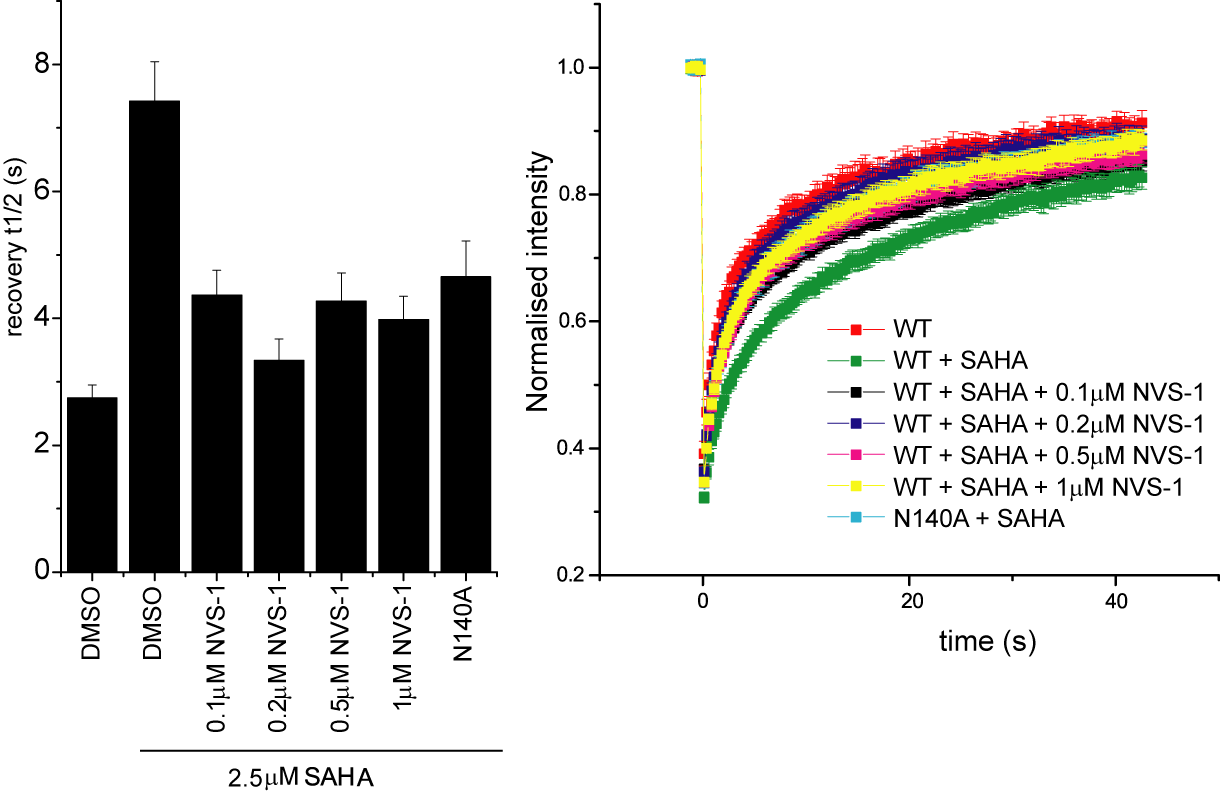
Half-times of fluorescence recovery (t1/2) after photo bleaching measured for CECR2 after treatment with NVS-CECR2-1 at different concentrations with or without SAHA.
Isothermal Titration Calorimetry (ITC)
All calorimetric experiments were performed on a VP-ITC micro-calorimeter (MicroCalTM, LLC Northampton, MA). Protein solutions were buffer exchanged by gel filtration or dialysis into buffer (20 mM Hepes pH 7.5, 150 mM NaCl, and 0.5 mM tris (2-carboxyethyl) phosphine (TCEP). All measurements were carried out at 288.15 K. All injections were performed using an initial injection of 2 µL followed by injections of 8 µL. The data were analysed with the MicroCal ORIGIN software package employing a single binding site model. The first data point was excluded from the analysis.
Temperature shift assay
Thermal melting experiments were carried out using a Stratagene Mx3005p Real Time PCR machine (Agilent Technologies). NVS-CECR2-1 was added at a final concentration of 10 µM. SYPRO Orange (Molecular Probes) was added as a fluorescence probe at a dilution of 1:1000 as described (6).
AlphaScreen Assay
Assays were performed as described previously with minor modifications (7). All reagents were diluted in 25 mM HEPES, 100 mM NaCl, 0.1 % BSA, pH 7.4 supplemented with 0.05 % CHAPS and allowed to equilibrate to room temperature prior to addition to plates. An 11-point 1:2.0 serial dilutions of the ligands was prepared on lowvolume 384-well plates (ProxiPlateTM-384 Plus, PerkinElmer, USA), using LabCyte Eco liquid handler. Plates filled with 5 µL of the assay buffer followed by 7 µL of peptide H4K5acK8acK12acK16ac. Plates were sealed and incubated for a further 30 minutes, before the addition of 8 µM of the mixture of streptavidin-coated donor beads (12.5 µg/mL) and nickel chelate acceptor beads (12.5 µg/mL) under low light conditions. Plates were foil-sealed to protect from light, incubated at room temperature for 60 minutes and read on a PHERAstar FS plate reader (BMG Labtech, Germany) using an AlphaScreen 680 excitation/570 emission filter set. IC50 values were calculated in Prism 5 (GraphPad Software, USA) after normalization against corresponding DMSO controls.
Fluorescence Recovery After Photobleaching (FRAP) Assay
FRAP studies were performed using U20S cells expressing full-length CECR2. Six hours after transfection 2.5 µM SAHA (to increase global histone acetylation) was added and cells were treated with 1 µM or 5 µM of NVS-CECR2-1 1 hour before imaging and half recovery times from the fluorescence signal of the bleached U2OS nuclei were plotted.
NanoBRET
U2OS cells were co-transfected with Histone H3.3-HaloTag and NanoLuc-CECR2. Twenty hours post-transfection cells were collected, washed with PBS, and exchanged into media containing phenol red-free DMEM and 4% FBS in the absence (control sample) or the presence (experimental sample) of 100 nM NanoBRET 618 fluorescent ligand (Promega). Cells were then treated with an increasing dose of NVS-CECR2-1. Five minutes prior to reading, NanoBRET furimazine substrate (Promega) was added to both control and experimental samples and plates were read on a CLARIOstar (BMG) equipped with 450/80 nm bandpass and 610 nm longpass filters with a 0.5 sec reading setting. A corrected BRET ratio was calculated and is defined as the ratio of the emission at 610 nm/450 nm for experimental samples (i.e. those treated with NanoBRET fluorescent ligand) subtracted by and the emission at 610 nm/450 nm for control samples (not treated with NanoBRET fluorescent ligand). BRET ratios are expressed as milliBRET units (mBU), where 1 mBU corresponds to the corrected BRET ratio multiplied by 1000. Relative IC50 values were estimated by non-linear regression analysis of (log) concentration of each inhibitor versus milliBRET ratios (GraphPad Prism).