| Probe | Negative control | |
 |
|  |
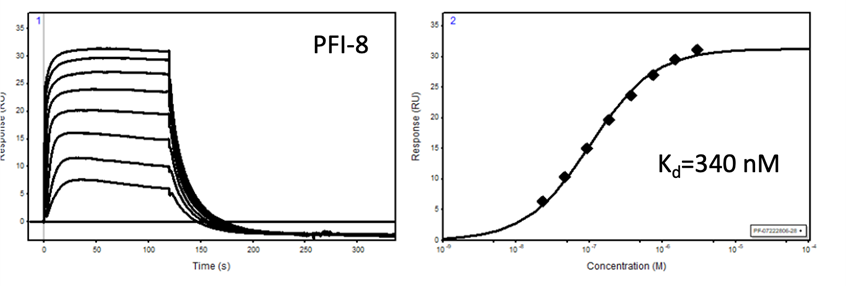
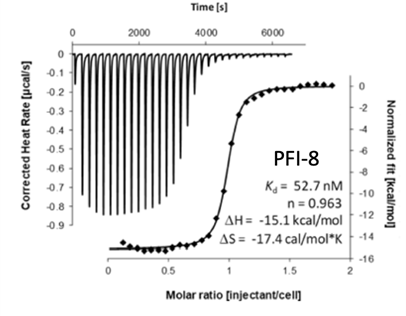
PFI-8 |
| PFI-8N |
The SGC in collaboration with Pfizer has discovered a potent chemical probe for the YEATS family, PFI-8. The YEATS-domain containing proteins are responsible for epigenetic signaling via acylated lysines including acetylation and higher order acylations such as propionylation, butyrylation, and crotonylation. PFI-8 binds YEATS 1-4 with FRET Kis of 495, 330, 462, and 33 nM respectively using a biotin-tagged crotonylated H3 peptide. PFI-8N, a negative control compound showed FRET Ki of >20 mM.
Binding of YEATS4 to PFI-8
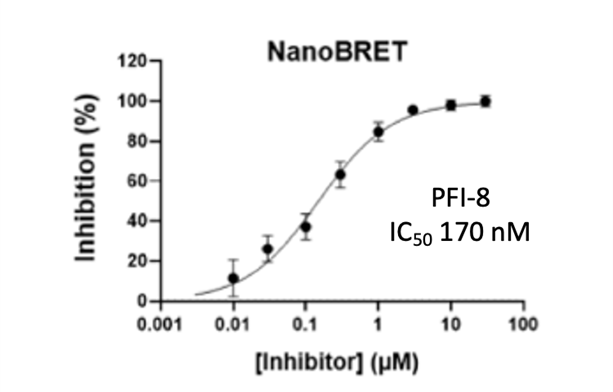
Cellular activity of PFI-8 using NanoBRET assay in HEK293 cells
1) (a) Heidenreich, D.; Moustakim, M.; Schmidt, J.; Merk, D.; Brennan, P. E.; Fedorov, O.; Chaikuad, A.; Knapp, S. Structure-based approach toward identification of inhibitory fragments for eleven-nineteen-leukemia protein (ENL). J. Med. Chem. 2018, 61, 10929−10934. (b) Moustakim, M.; Christott, T.; Monteiro, O. P.; Bennett, J.; Giroud, C.; Ward, J.; Rogers, C. M.; Smith, P.; Panagakou, I.; Diaz-Saez, L.; Felce, S. L.; Gamble, V.; Gileadi, C.; Halidi, N.; Heidenreich, D.; Chaikuad, A.; Knapp, S.; Huber, K. V. M.; Farnie, G. Heer, J.; Manevski, N.; Poda, G.; Al-Awar, R.; Dixon, D. J.; Brennan, P. E.; Fedorov, O. Discovery of an MLLT1/3 YEATS domain chemical probe. Angew. Chem., Int. Ed. 2018, 57, 16302−16307. (c) Christott, T.; Bennett, J.; Coxon, C.; Monteiro, O.; Giroud, C.;Beke, V.; Felce, S. L.; Gamble, V.; Gileadi, C.; Poda, G.; Al-Awar, R.;Farnie, G.; Fedorov, O. Discovery of a Selective Inhibitor for the YEATS Domains of ENL/AF9. SLAS Discov. 2019, 24, 133−141. (d) Ni, X.; Heidenreich, D.; Christott, T.; Bennett, J.; Moustakim, M.; Brennan, P. E., Fedorov, O.; Knapp, S.; Chaikuad, A. Structural insights into interaction mechanisms of alternative piperazine-urea YEATS domain binders in MLLT1. ACS Med. Chem. Lett. 2019, 10, 1661−1666. (e) Structural Genomics Consortium (SGC): PFI-6 A novel chemical probe for MLLT1/3. https://www.thesgc.org/chemicalprobes/PFI-6 (accessed June 23, 2020). (f) Structural Genomics Consortium (SGC): NVS-MLLT-1 A Potent and Selective inhibitor of YEATS proteins. https://www.thesgc.org/chemical-probes/NVS-MLLT-1 (accessed March 5, 2020) (g) Asiaban, J. N.; Milosevich, N.; Chen, E.; Bishop, T. R.; Wang, J.; Zhang, Y.; Ackerman, C. J.; Hampton, E. N.; Young, T. S.; Hull, M. V., Cravatt, B. F., Erb, M. A. Cell-based ligand discovery for the ENL YEATS domain. ACS Chem. Biol. 2020, 15, 895–903. (h) Ni, X.; Londregan, A. T.; Owen, D. R.; Knapp, S.; Chaikuad, A. ACS Chem. Bio. Structure and Inhibitor Binding Characterization of Oncogenic MLLT1 Mutants. 2021, 16, 569-576. (i) Listunov, D.; Linhares, B. M.; Kim, E.; Winkler, A.; Simes, M. L.; Weaver, S.; Cho, H. J.; Rizo, A.; Zolov, S.; Venkateshwar G. K.; Grembecka, J.; Cierpicki, T. Development of potent dimeric inhibitors of GAS41 YEATS domain. Cell Chem.Bio. 2021, 28, 1716-1727.
2) Measurement of ATP-depletion over 72 hours: Green, N.; Aleo, M. D.; Louise-May, S.; Price, D. A.; Will, Y. Using an in vitro cytotoxicity assay to aid in compound selection for in vivo safety studies. Bioorg. Med. Chem. Lett. 2010, 20, 5308-5312.
The probe and control may be requested here.
| Probe | Negative control | |
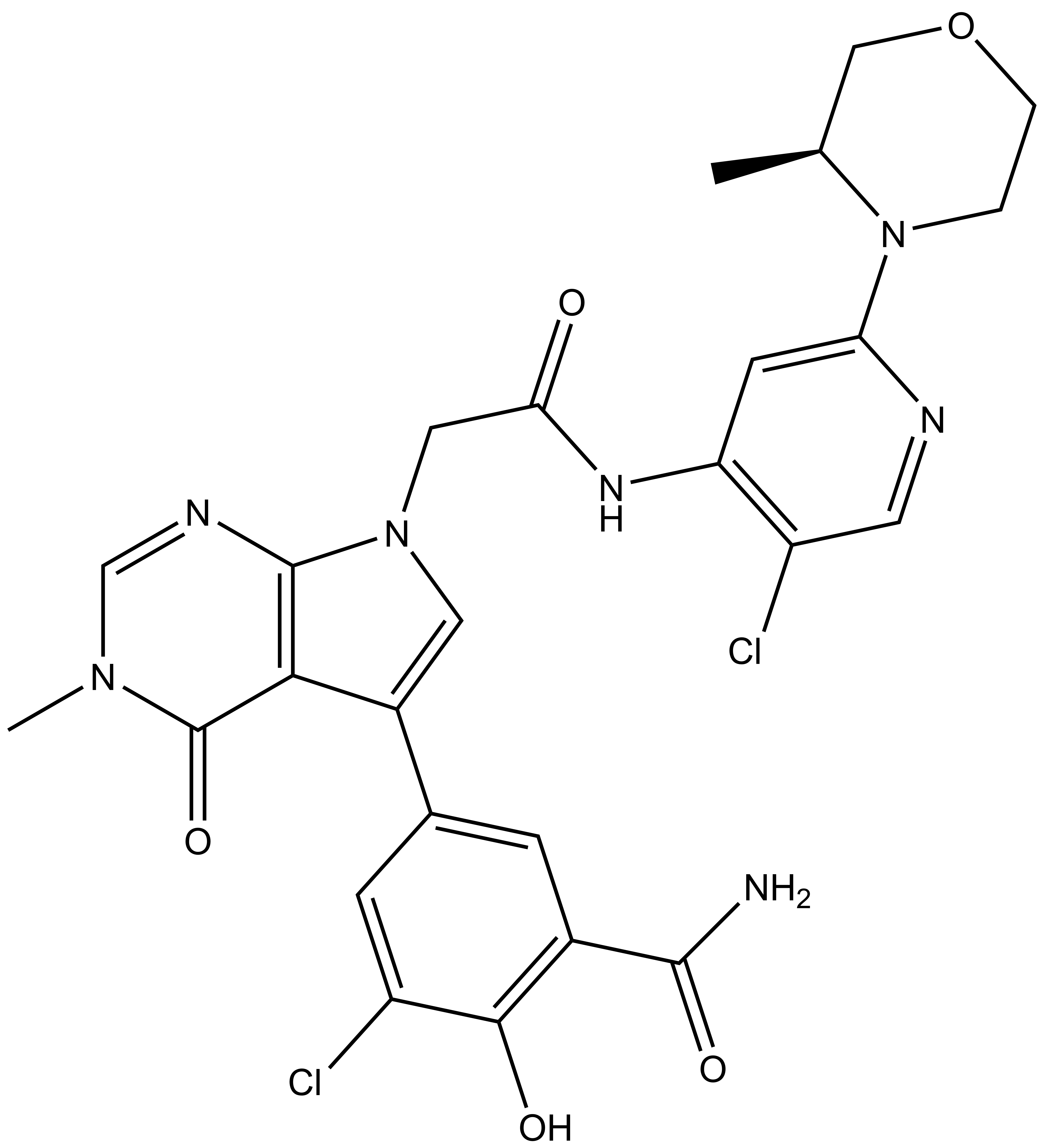 |
| 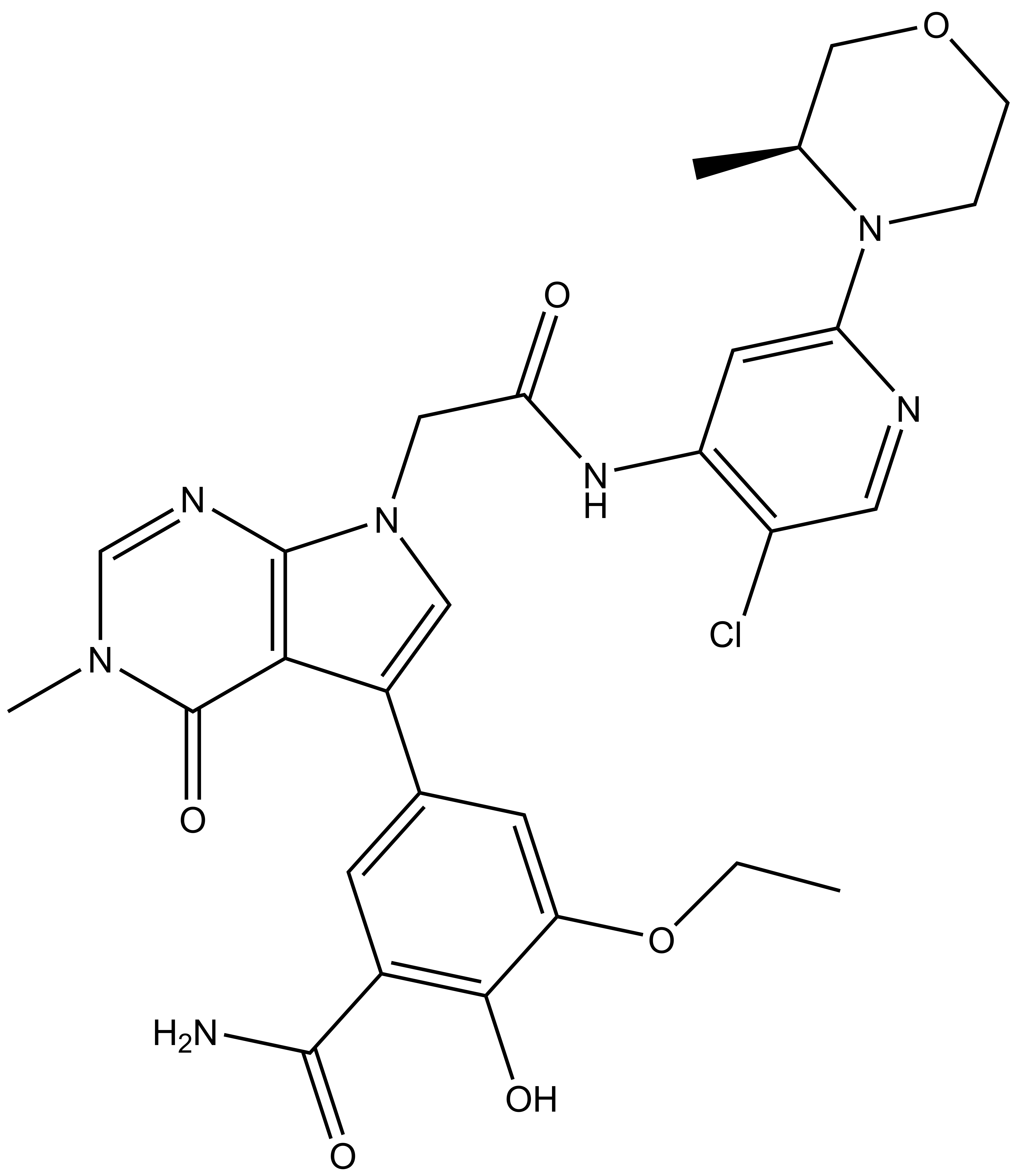 |
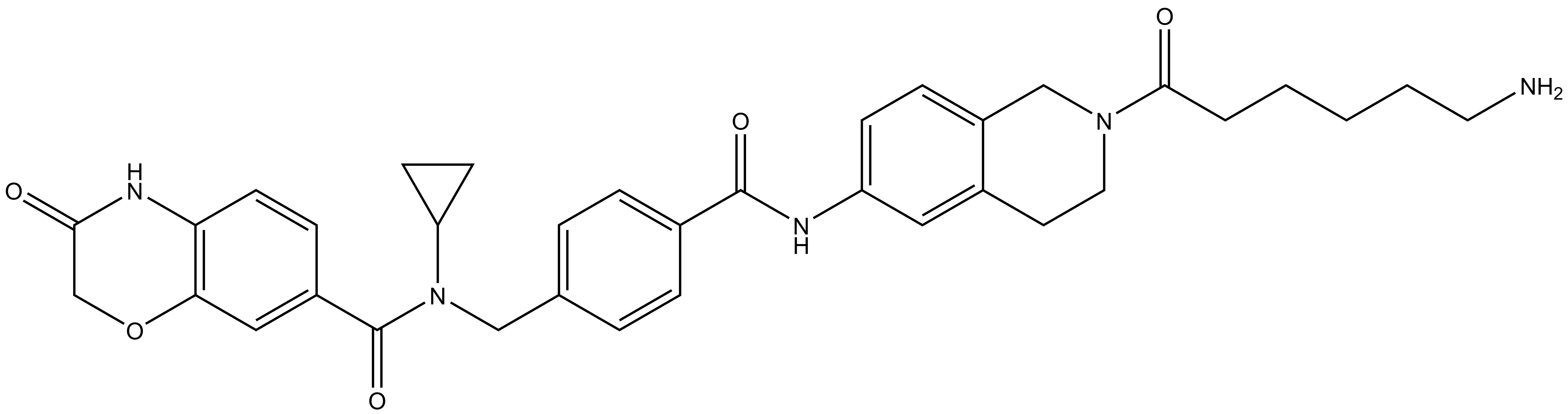
OICR11029 |
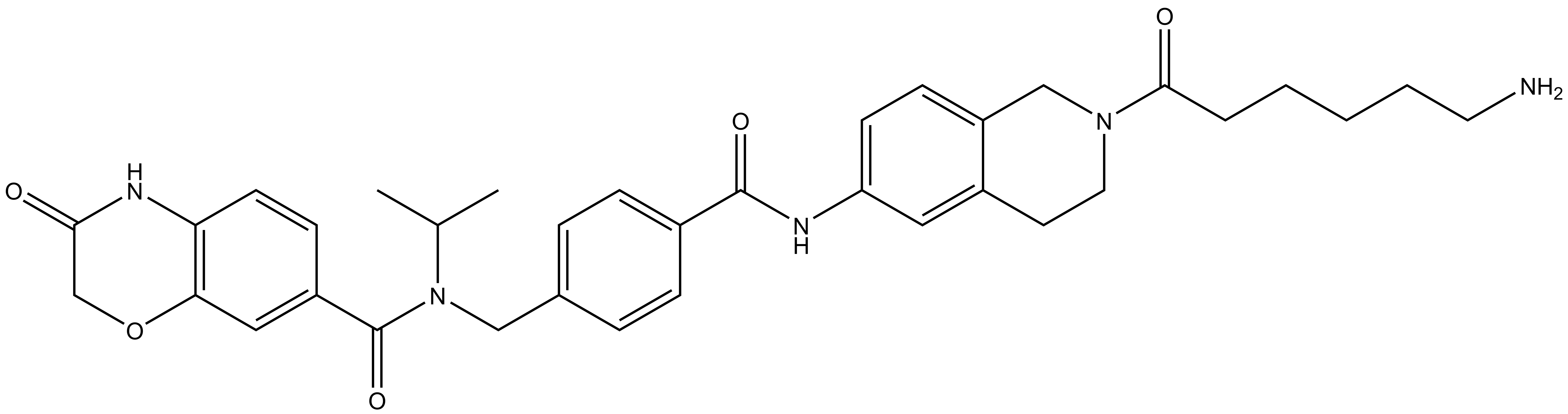
| OICR11600 |
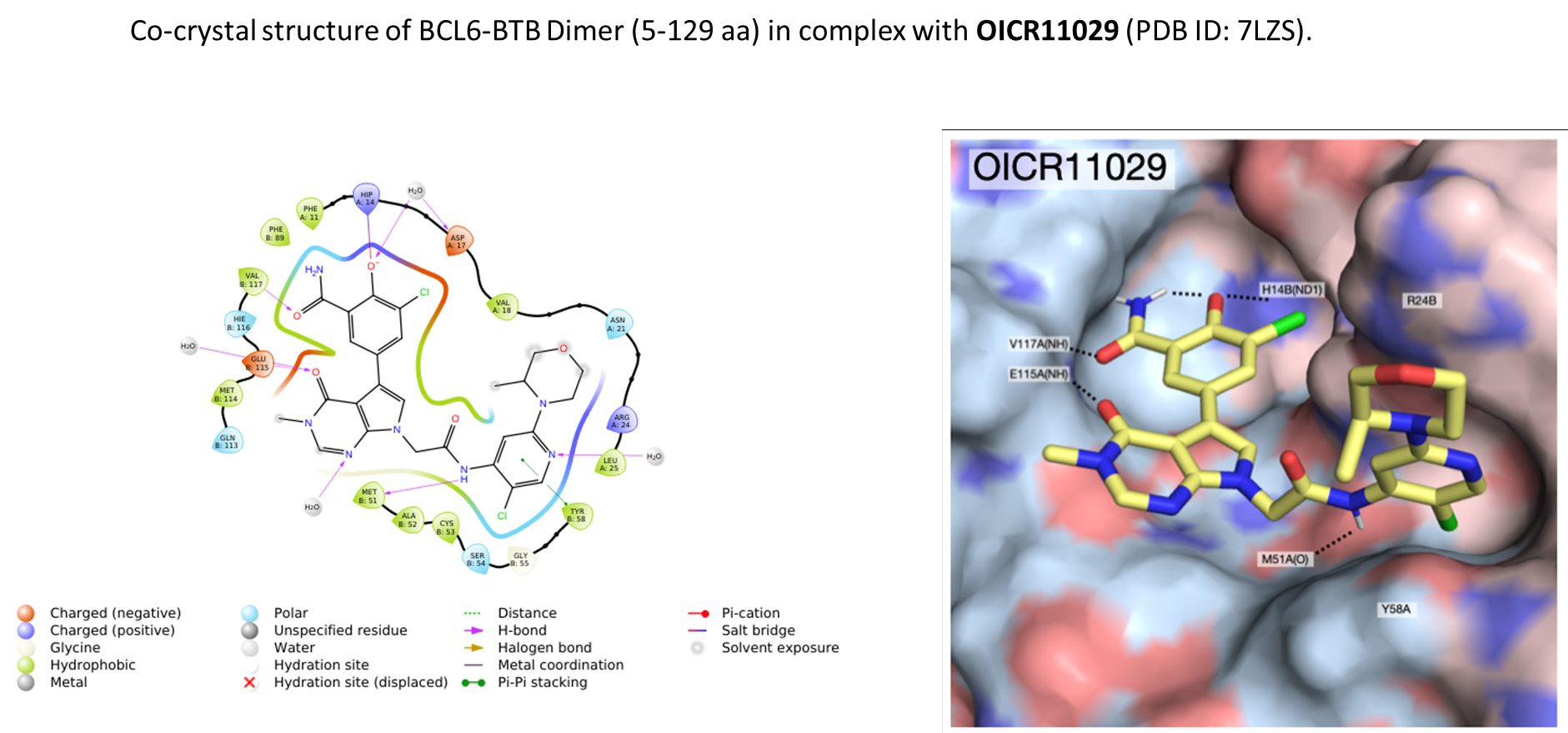
B cell lymphoma 6 (BCL6) is a highly regulated transcriptional repressor critical for the development and maintenance of germinal centers (GCs). BCL6 is required for generation of an effective humoral immune response and misexpression of BCL6 is widely seen in several forms of non-Hodgkin Lymphoma (NHL). B cell lymphoma 6 (BCL6) is a member of the BTB-ZF family of transcription factors. In these proteins, the N-terminal BTB domain binds to epigenetic modifiers (co-repressors) through protein-protein interactions, while the C-terminal zinc fingers mediate site-specific DNA binding [1, 2]. These corepressor proteins use a 17 amino acid motifs to bind to a solvent-exposed “lateral groove” formed by the two chains of the BCL6 BTB homodimer. Agents that bind to the BCL6 BTB lateral groove compete for corepressor binding and can reverse the repression activities of BCL6 [3]. In principle, the selective targeting of protein-protein interactions (PPI’s) in the BCL6 BTB domain is a more precise form of inhibition of BCL6 relative to complete inactivation or removal of the protein. OICR in collaboration with the UHN has developed, a potent, and highly selective BCL6-BTB chemical probe [4]. OICR11029 shows high in vitro as well as cellular potency. OICR11029 is accompanied by a negative control (OICR11600), which is structurally closely related to the probe molecule.
| Probe | Negative control | |
|
| |
OICR11029 |
| OICR11600 |
| Physical and Chemical Properties of OICR11029 | |
Molecular Weight | 586.40 |
Molecular Formula | C26H25N7O5Cl2 |
| IUPAC Name | (S)-3-chloro-5-(7-(2-((5-chloro-2-(3-methylmorpholino)pyridin-4-yl)amino)-2-oxoethyl)-3-methyl-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)-2-hydroxybenzamide |
| cLogP | 2.04 |
| tPSA | 153.16 |
No. of Chiral Centers | 1 |
No. of rotatable bonds | 6 |
No. of H-bond Acceptors | 8 |
No. of H-bond Donors | 3 |
| Storage | room temperature |
| Solubility (Kinetic) | 29 uM |
SMILES: CN1C=NC2=C(C(C3=CC(C(N)=O)=C(C(Cl)=C3)O)=CN2CC(NC4=C(C=NC(N5CCOC[C@@H]5C)=C4)Cl)=O)C1=O
InChI=1S/C26H25Cl2N7O5/c1-13-11-40-4-3-35(13)20-7-19(18(28)8-30-20)32-21(36)10-34-9-16(22-25(34)31-12-33(2)26(22)39)14-5-15(24(29)38)23(37)17(27)6-14/h5-9,12-13,37H,3-4,10-11H2,1-2H3,(H2,29,38)(H,30,32,36)/t13-/m0/s1
InChlKey: NDRFFOKFYGUOKD-ZDUSSCGKSA-N
| Physical and Chemical Properties of OICR11600 | |
Molecular Weight | 596.03 |
Molecular Formula | C28H30ClN7O6
|
| IUPAC Name | (S)-5-(7-(2-((5-chloro-2-(3-methylmorpholino)pyridin-4-yl)amino)-2-oxoethyl)-3-methyl-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)-3-ethoxy-2-hydroxybenzamide |
| cLogP | 1.84 |
| tPSA | 162.39 |
No. of Chiral Centers | 1 |
No. of rotatable bonds | 8 |
No. of H-bond Acceptors | 9 |
No. of H-bond Donors | 3 |
| Storage | room temperature |
| Solubility (Kinetic) | Not tested |
SMILES: CN1C=NC2=C(C(C3=CC(C(N)=O)=C(C(OCC)=C3)O)=CN2CC(NC4=C(C=NC(N5CCOC[C@@H]5C)=C4)Cl)=O)C1=O
InChI=InChI=1S/C28H30ClN7O6/c1-4-42-21-8-16(7-17(25(21)38)26(30)39)18-11-35(27-24(18)28(40)34(3)14-32-27)12-23(37)33-20-9-22(31-10-19(20)29)36-5-6-41-13-15(36)2/h7-11,14-15,38H,4-6,12-13H2,1-3H3,(H2,30,39)(H,31,33,37)/t15-/m0/s1
InChlKey: PWMORLQFVWCZTJ-HNNXBMFYSA-N
OICR11029 was found to be highly selective as determined at Eurofins Pharma in their kinome-wide panel at 3 µM and their CEREP safety panel at 10 uM assay.
OICR11029 was assessed in the Eurofins CEREP Safety panel of at 10 µM concentration; Only 1/53 >50%; MT1 agonist inhibition at 10 µM
| BTB domain | OICR11029 (SPR KD, uM) |
| BCL6 | 0.010 |
| BAZF (BCL6B) | 1.40 |
| FAZF, PLZF, Miz1, Kaiso, LRF | >10 |
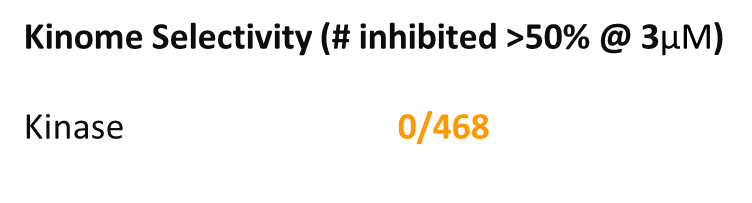
OICR11029 |
OICR11600 | Fold over negative control | |
| Potency | |||
| KD_SPR [IC50, µM] | 0.010 | 41.1 | X 4,110 |
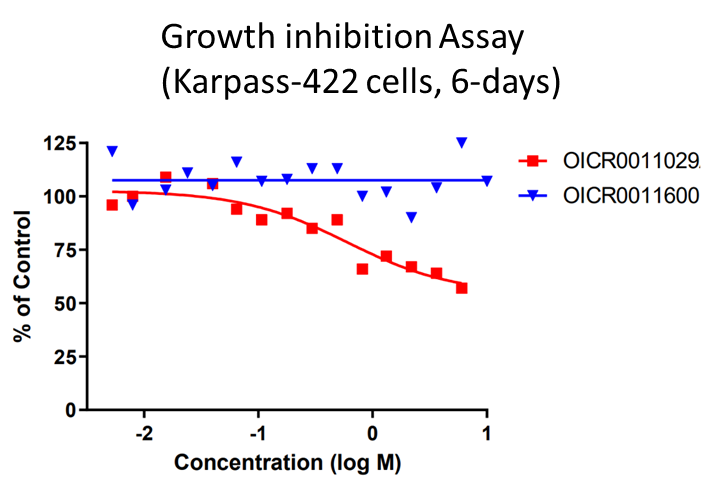
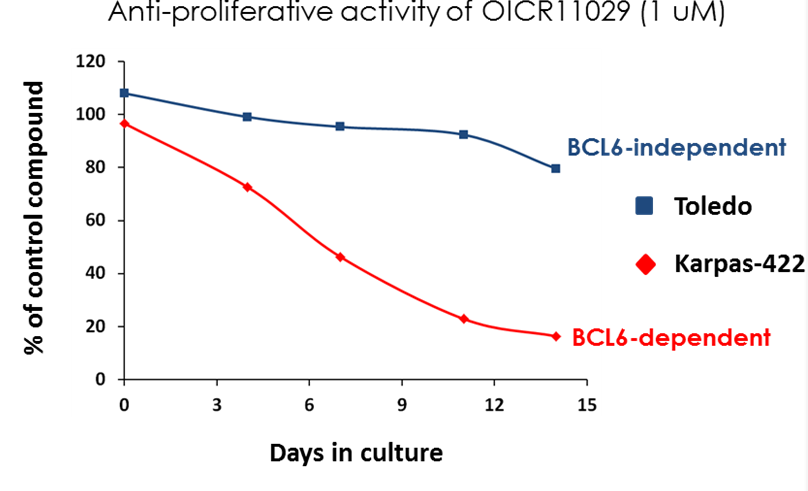
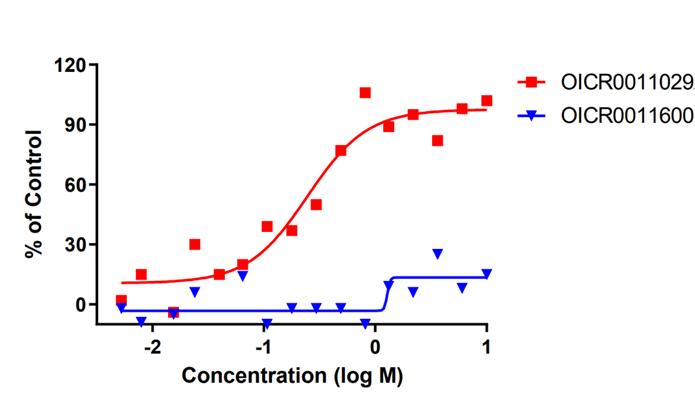
OICR11029 displayed an IC50 of 375 nM in the cellular luciferase assay in SUDHL4 cells and 602 nM in the Karpass-422 short term growth inhibition assay. NB: Karpass-422 is a BCL6-dependent cancer cell line.
1.Ahmad, K.F. et al., Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol Cell, 2003, 12 (6): 1551-64.
2.Ghetu, A., et al., Structure of a BCOR corepressor peptide in complex with the BCL6 BTB domain dimer, Mol Cell. 2008; 29(3): 384–391.
3.Cerchietti, L.C., et al., A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell, 2010, 17 (4): p. 400-11.
4.Mamai, A. et al., ACS Med. Chem. Lett. 2023, 14, 2, 199–210
| Probe | Negative control | |
 |
|  |
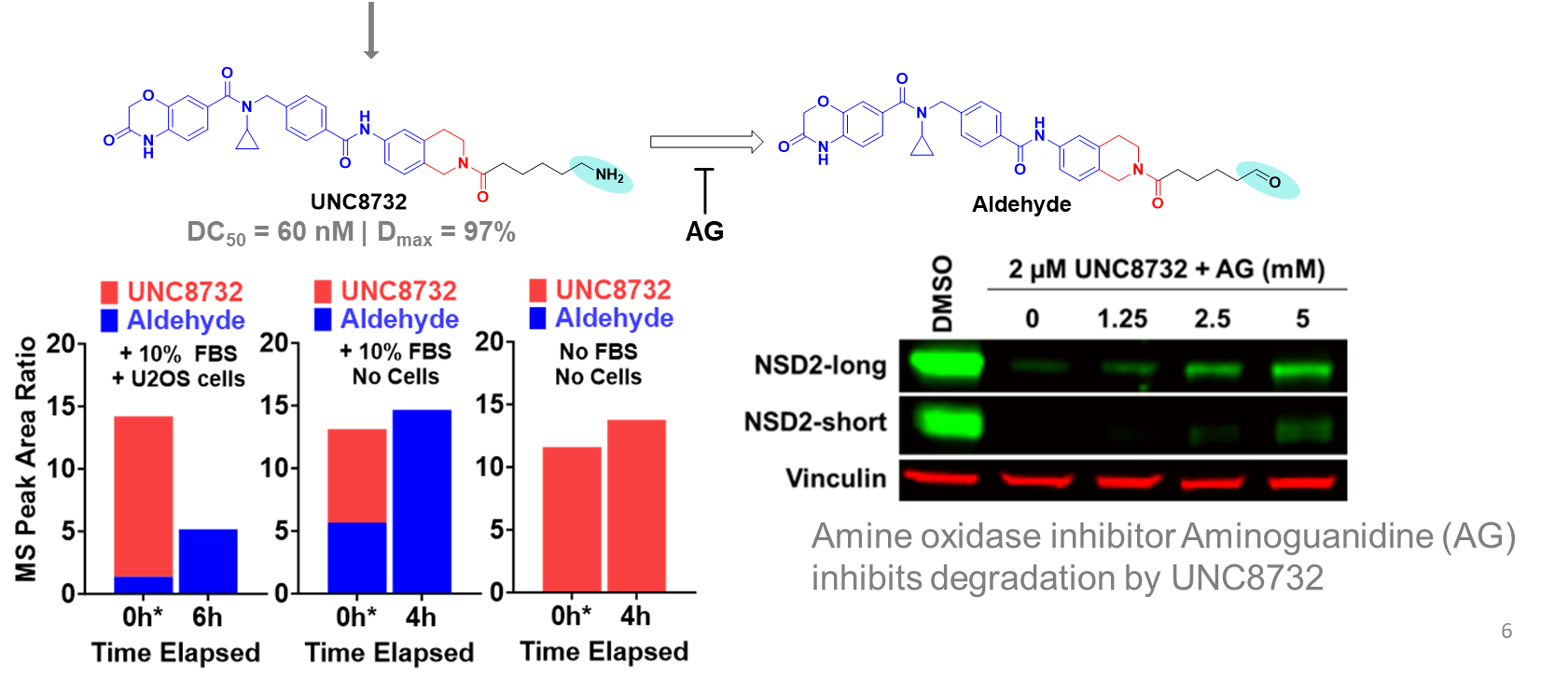
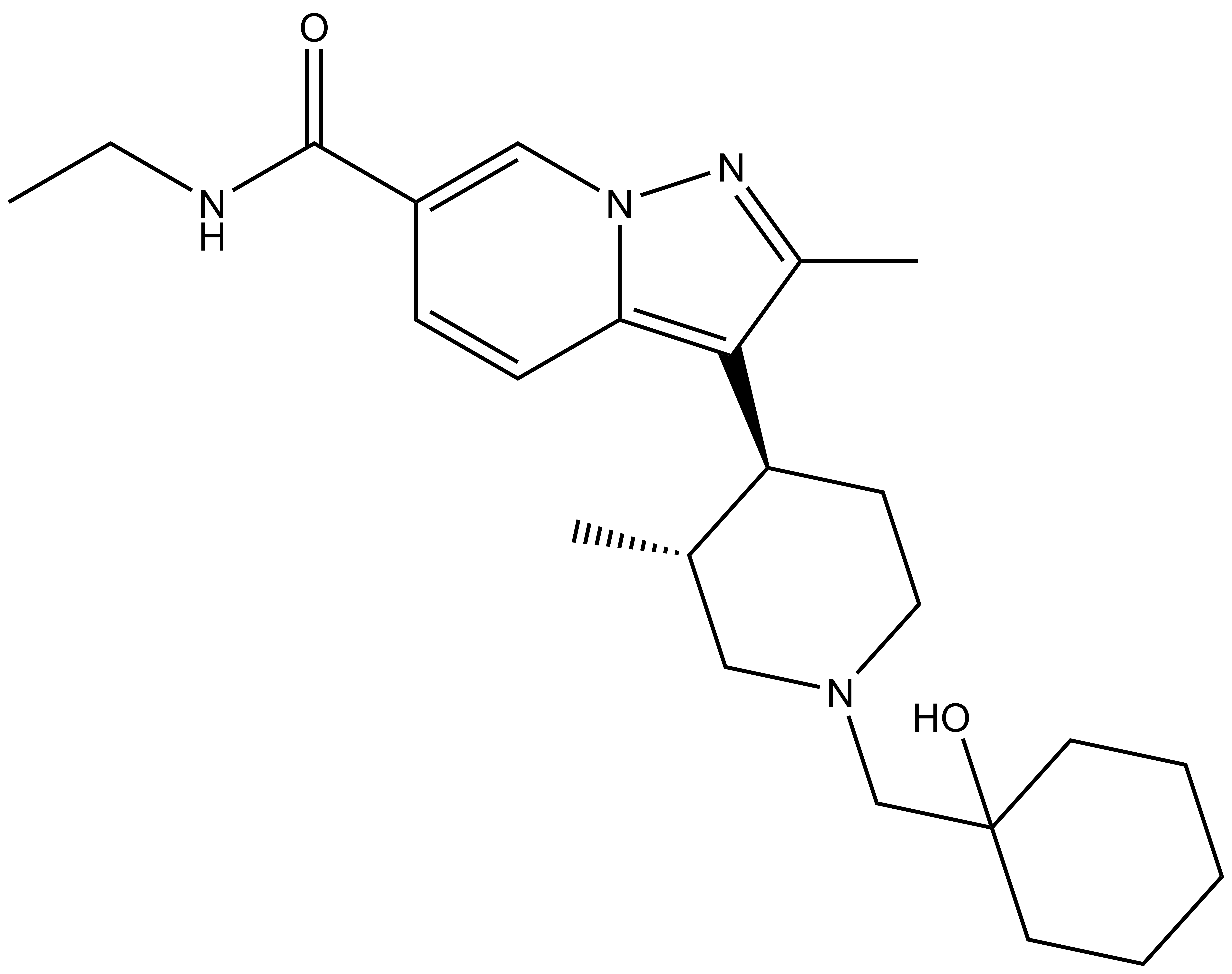
UNC8732 |
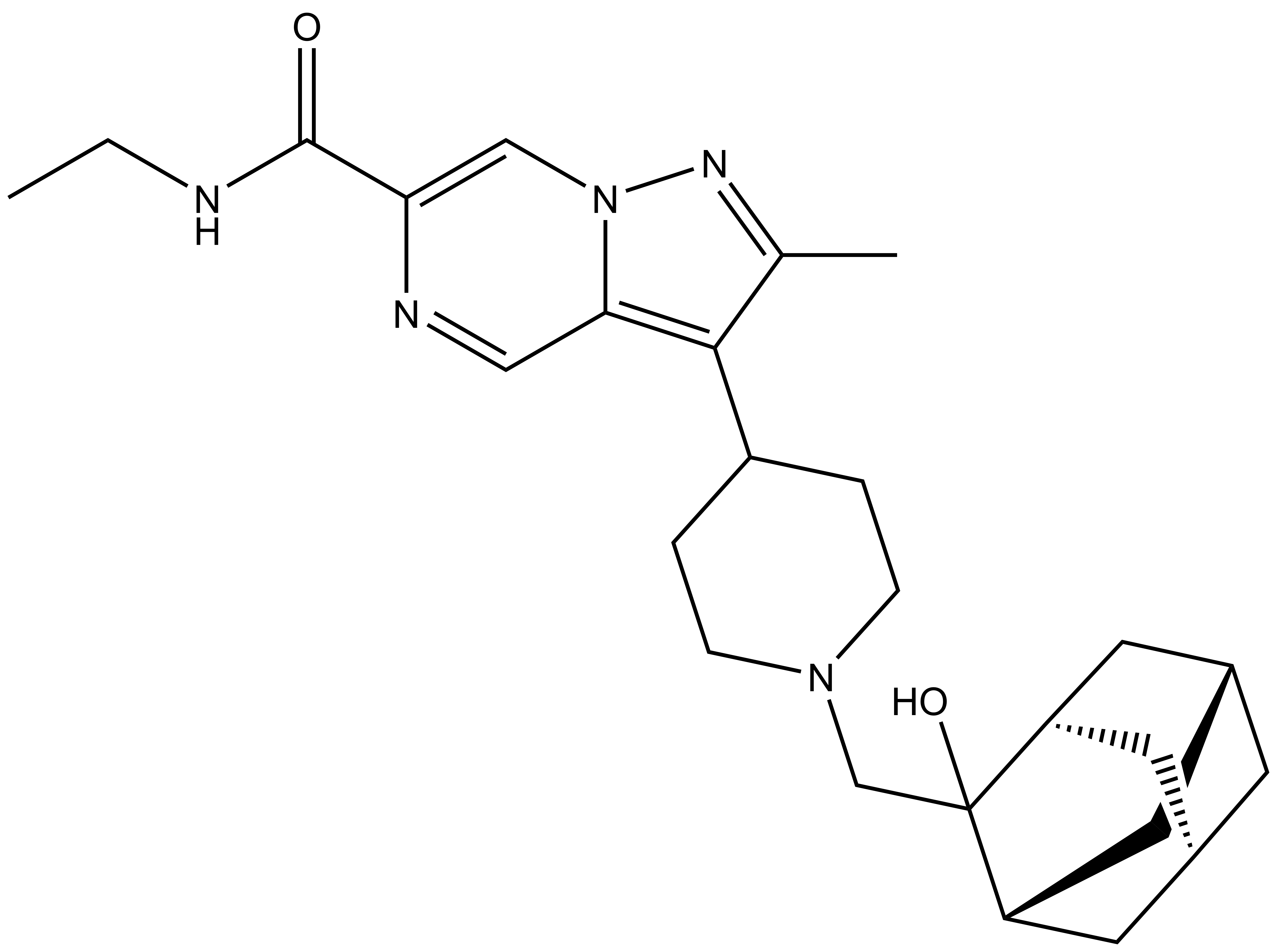
| UNC8884 |