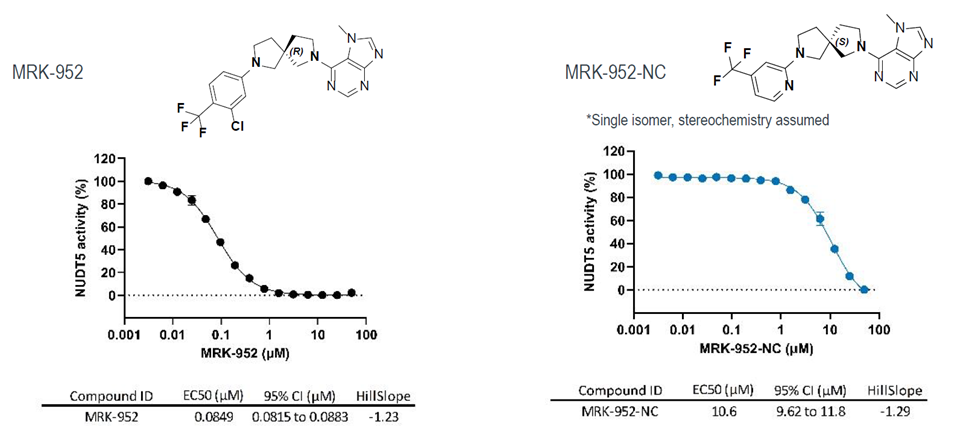
| Probe | | Negative control |
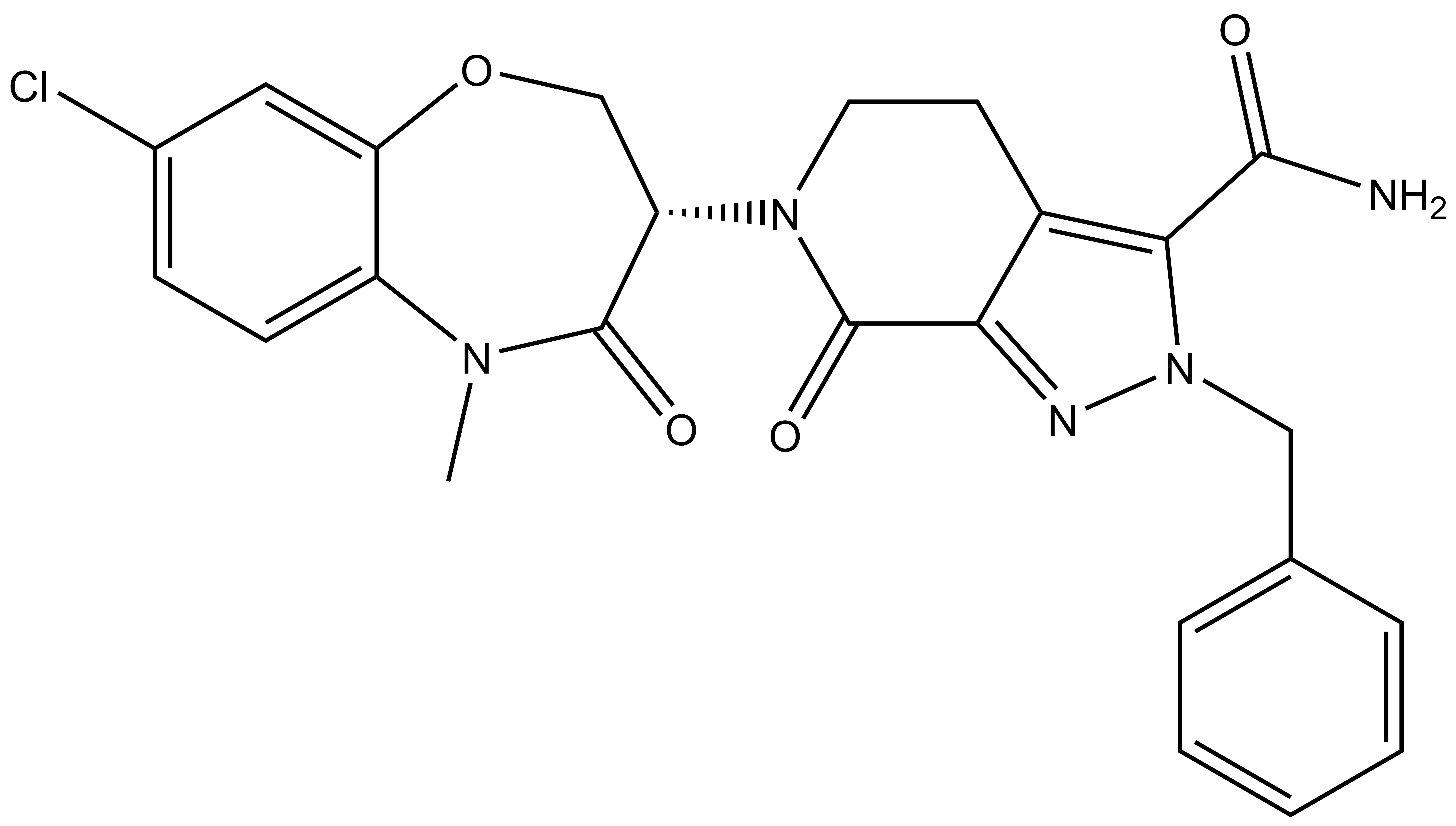 | | 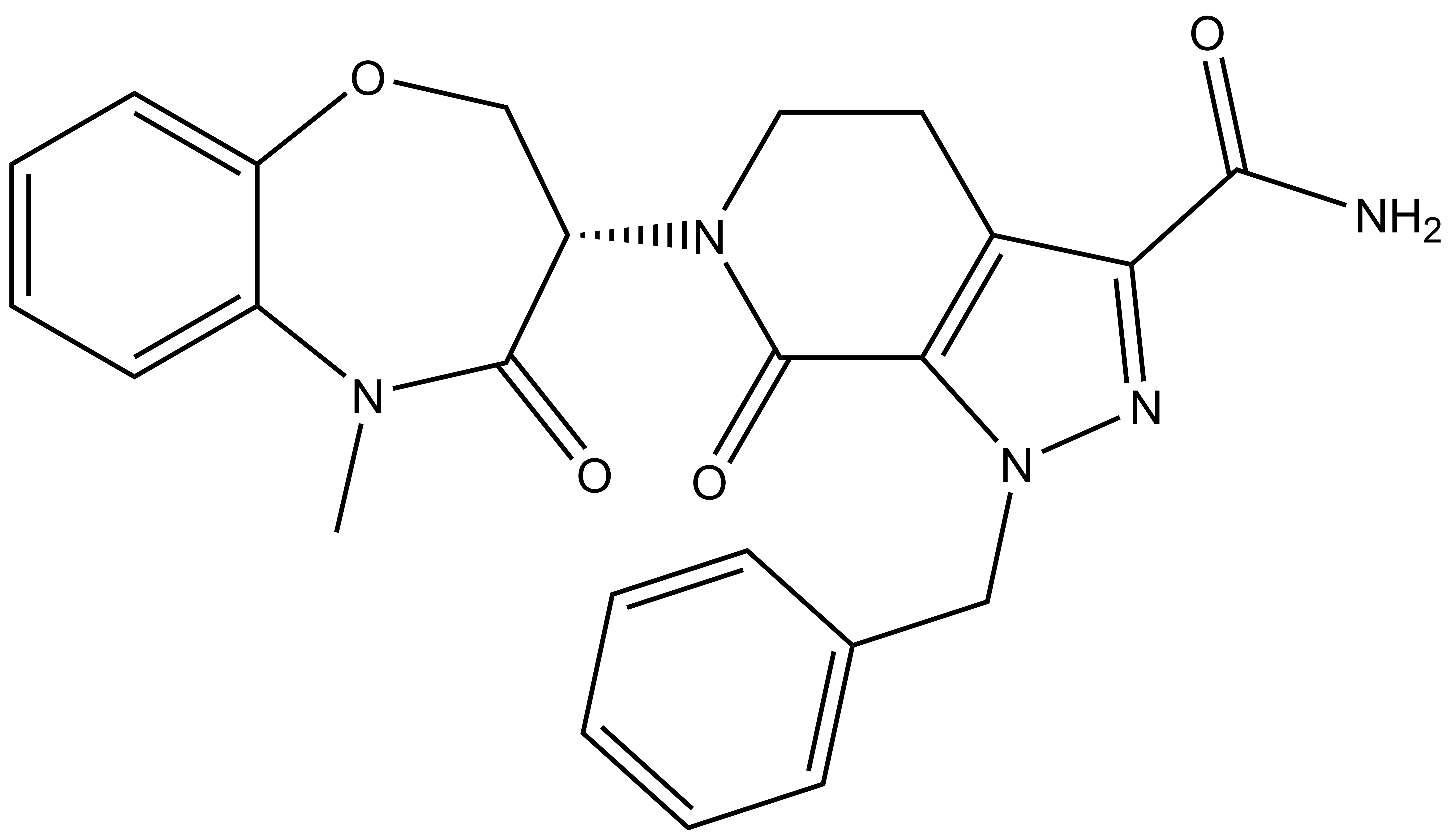 |
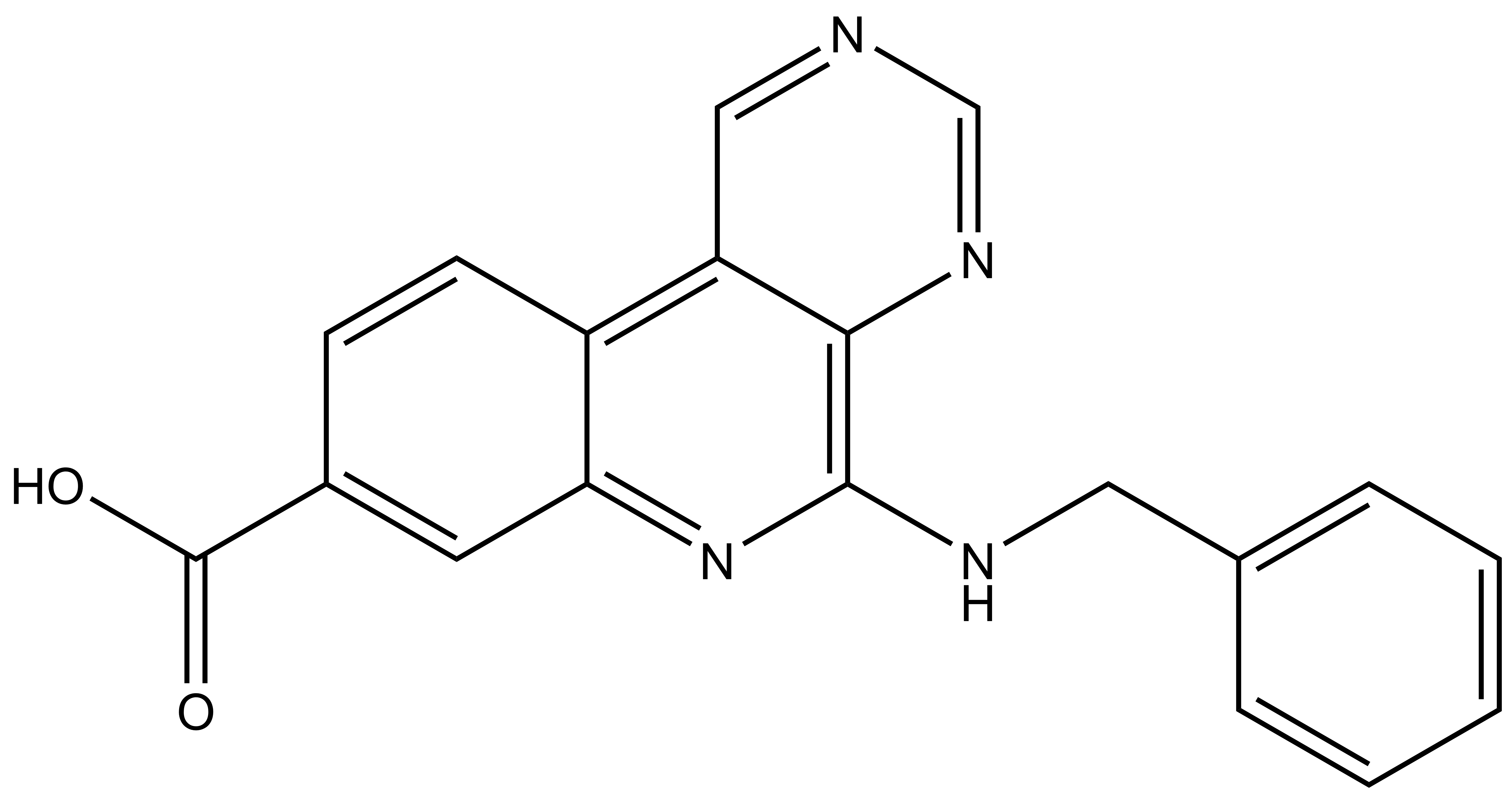
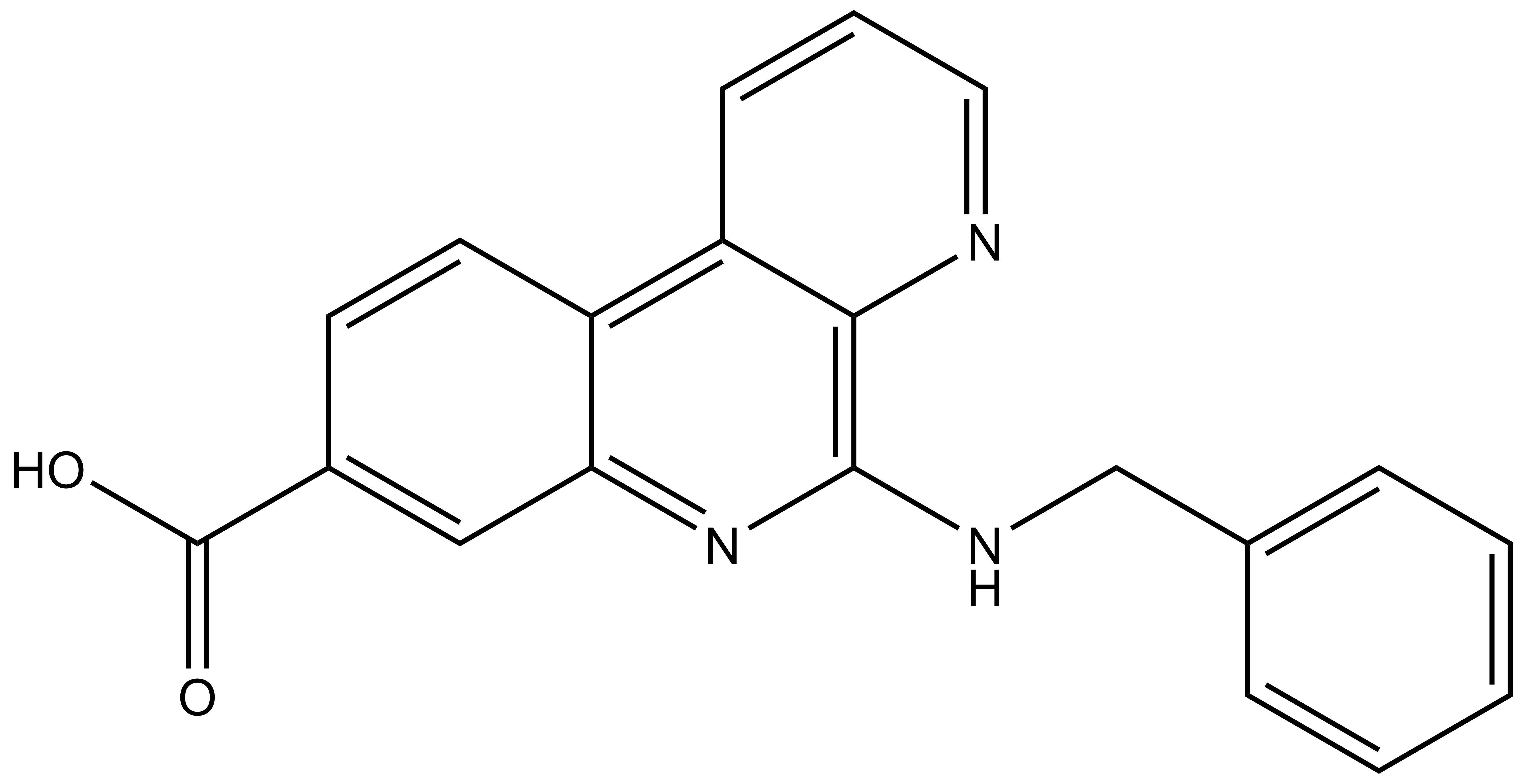
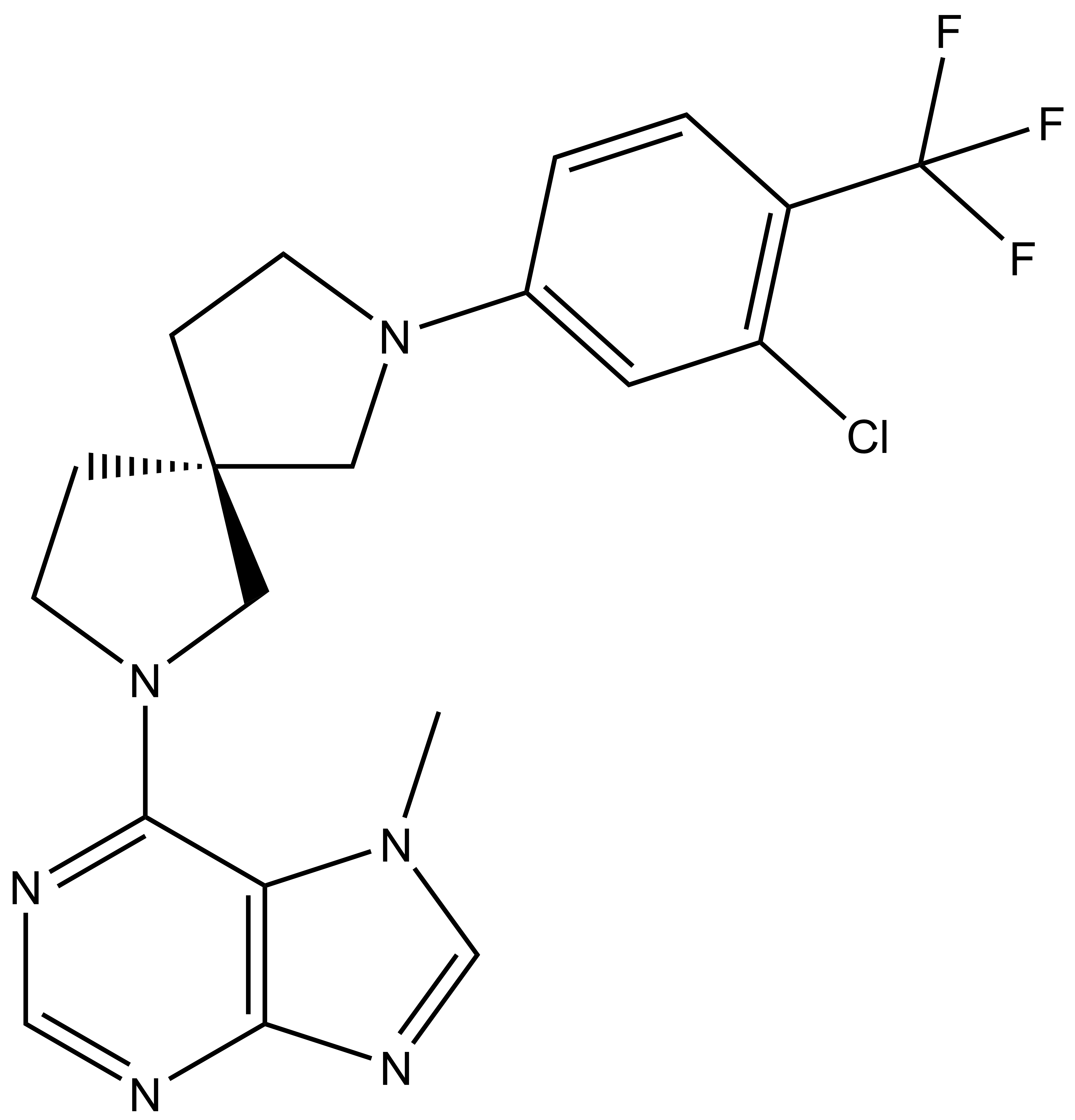
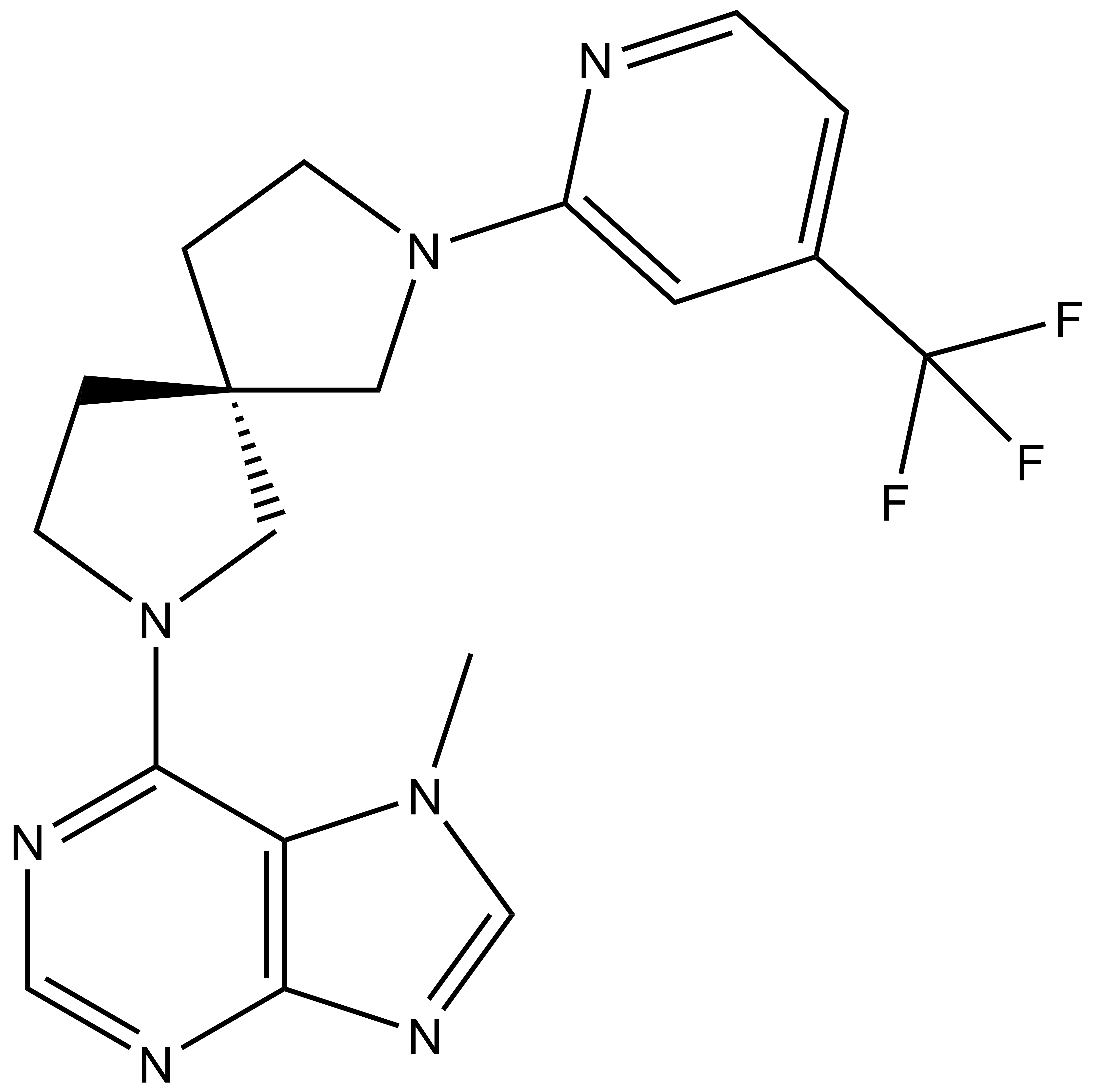
LIJTF500025 | | LIJTF500120 |
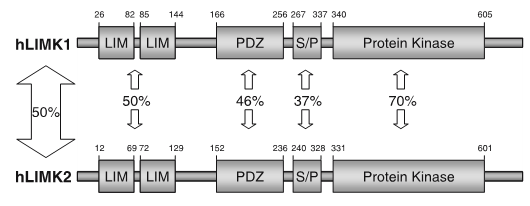
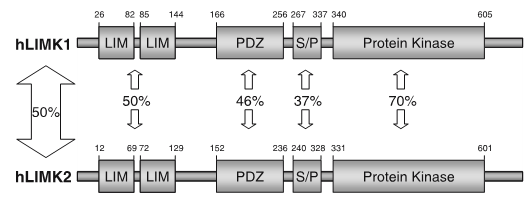
LIM kinases belong to the family of cytoplasmic tyrosine-like kinases with dual specificity (serine/threonine and tyrosine). However, known LIMK substrate are usually phosphorylated at serine and threonine residues LIM kinases comprises LIM kinase 1 (LIMK1) and LIM kinase 2 (LIMK2) which show 50% sequence identity in human. Both LIMK1 and LIMK2 present with a unique domain organization containing two N-terminal LIM domains, a PDZ domain, a proline/serine-rich domain and a C-terminal kinase domain [1].
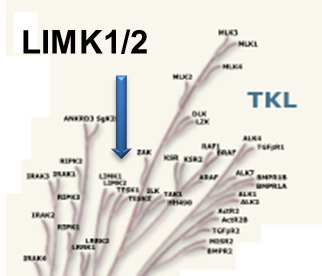
Both proteins are expressed widely in embryonic and adult tissues, but show some cell-type specific expression. Accordingly, the two kinases have overlapping functions, but appear non-redundant. Knockout studies in mice show that LIMK1 is required for development of the central nervous system [2], whereas LIMK2 knockout impairs the activity of testicular germ cells [3].
LIMKs are effectors of cell morphology and motility and apoptosis by regulating the actin cytoskeleton. The LIMKs signal downstream from Rho GTPases and are activated by phosphorylation of the activation loop by upstream kinases, including Rho kinase (ROCK), PAK1/2/4 and MRCKα. The best characterized LIMK substrates are cofilin1 (non-muscle cofilin), cofilin2 (muscle cofilin) and destrin (actin depolymerizing factor, ADF). Phosphorylation of cofilin serine-3 inactivates the actin severing ability promoting F-actin polymerization, stress fibre formation and focal adhesion formation [4].
LIM kinases can shuttle between the cytoplasm and the nuclear compartment of a cell, a process tightly regulated by association with other partners such as p57kip2 and phosphorylation in the activation segment by PAK kinases [1]. Inhibition of LIMK hyper-stabilizes mitotic spindles inducing a G2/M cell cycle block suggesting an important role for these kinases in microtubule dynamics [5].
Increased phosphorylation of LIMK1 has been reported in neurons in areas affected with Alzheimer Disease [6]. LIM kinases play important roles in cancer metastasis like highly invasive prostate and breast cancer, which is reversed by gene silencing [7, 8]. LIMK1 overexpression is also found in malignant melanoma, as well as most tumour cell lines. Other applications for LIMK inhibitors are open-angle glaucoma [9]. In addition, LIMK1 interacts with the long isoform of the type II bonemorphogenetic protein (BMP) receptor contributing to the pathology of Fragile X syndrome, a common inherited form of intellectual disability [10].
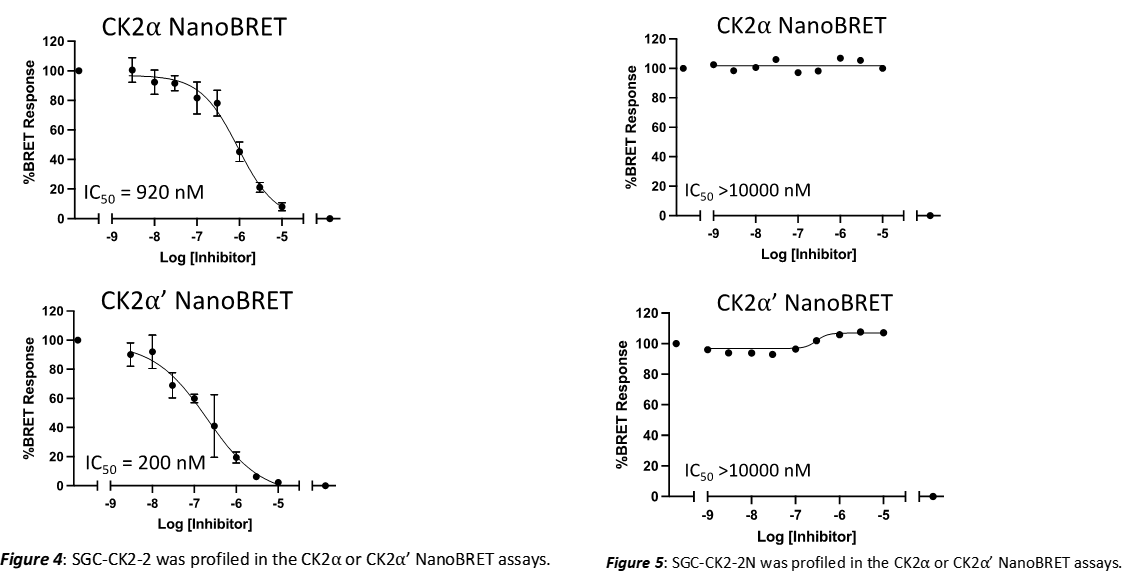
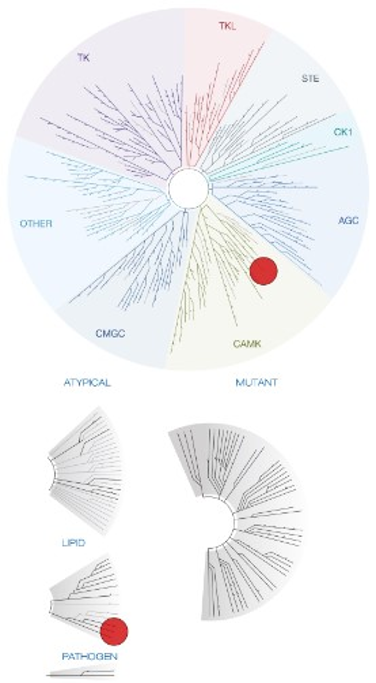
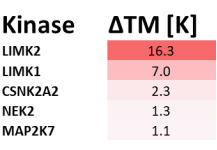
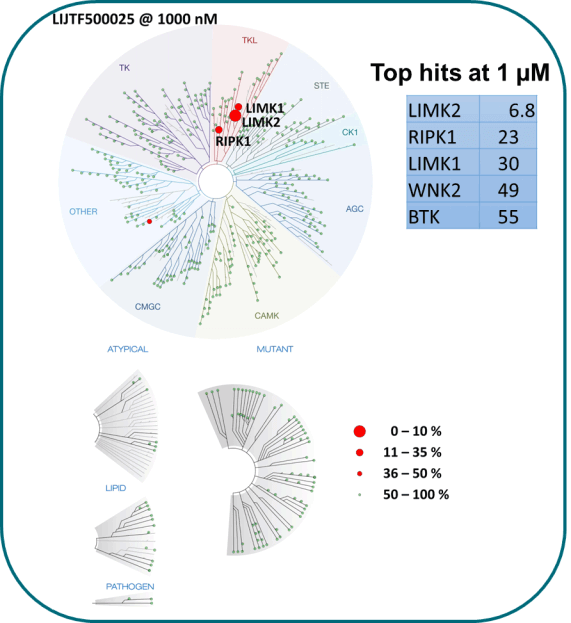
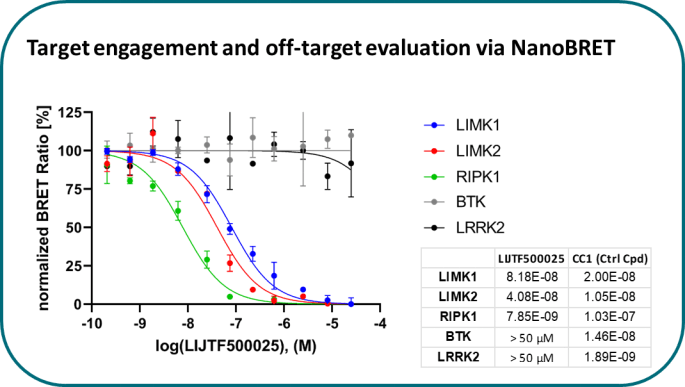
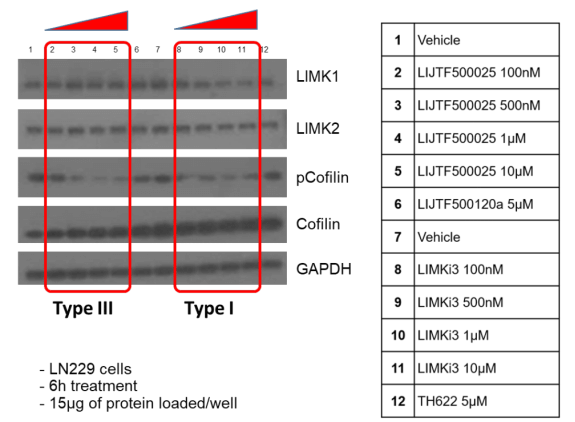
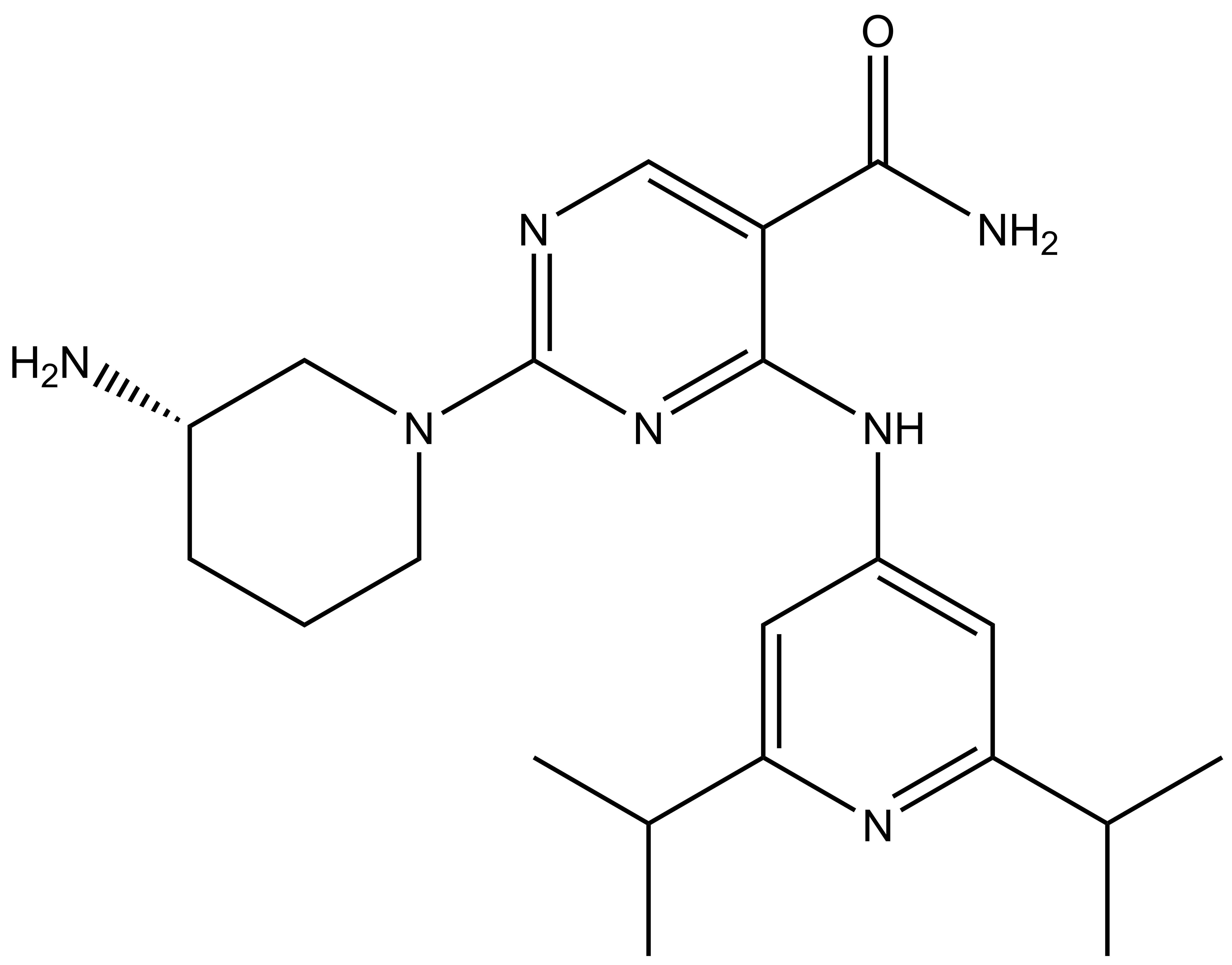
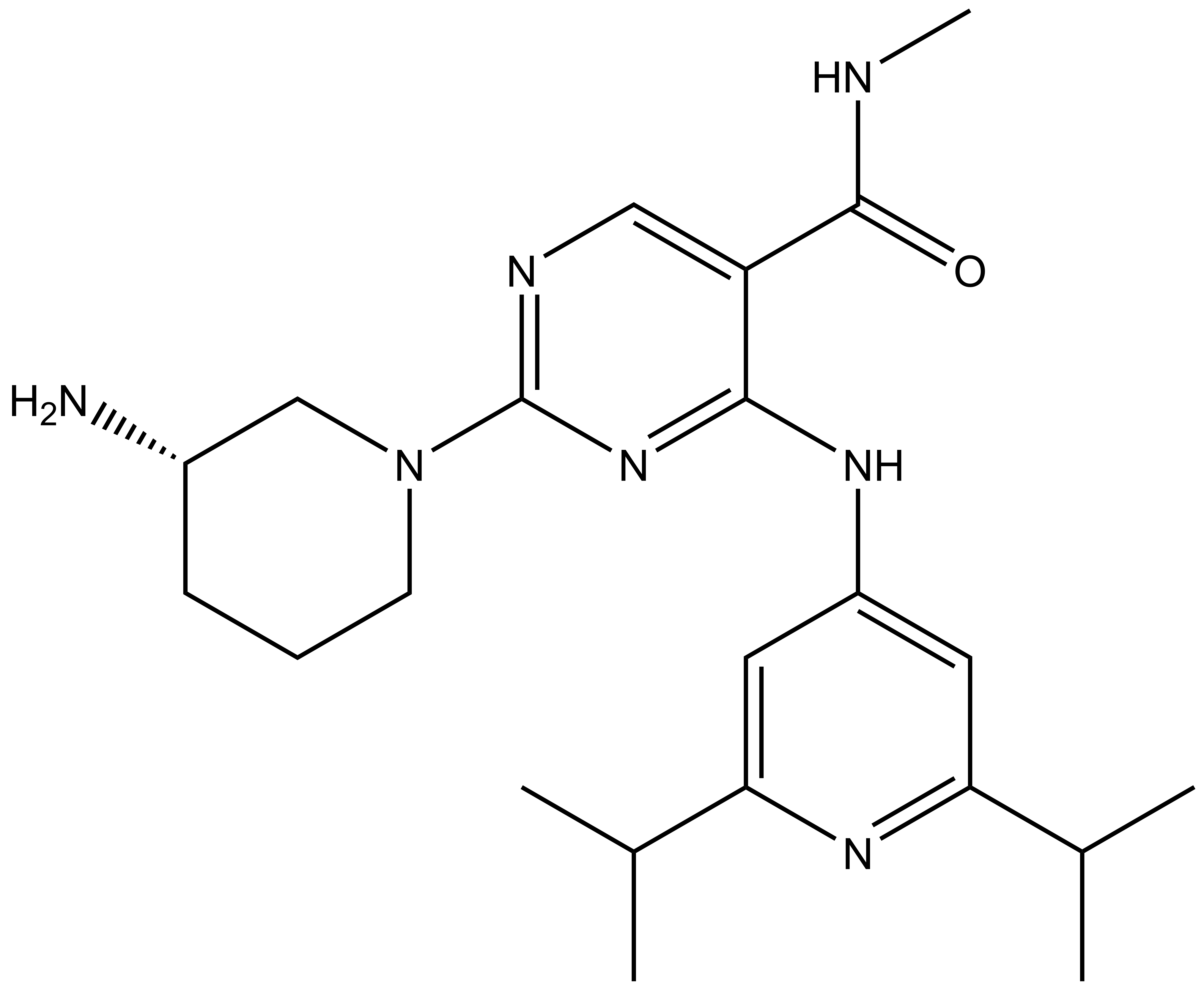
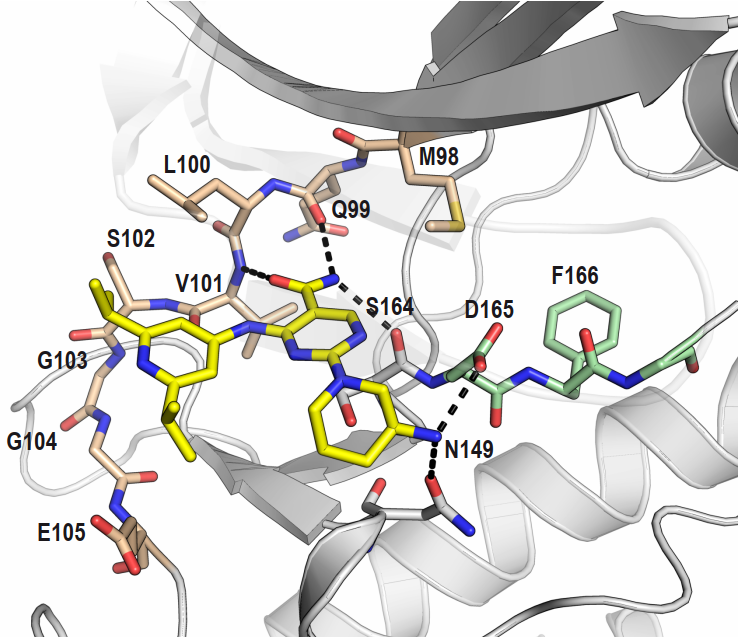
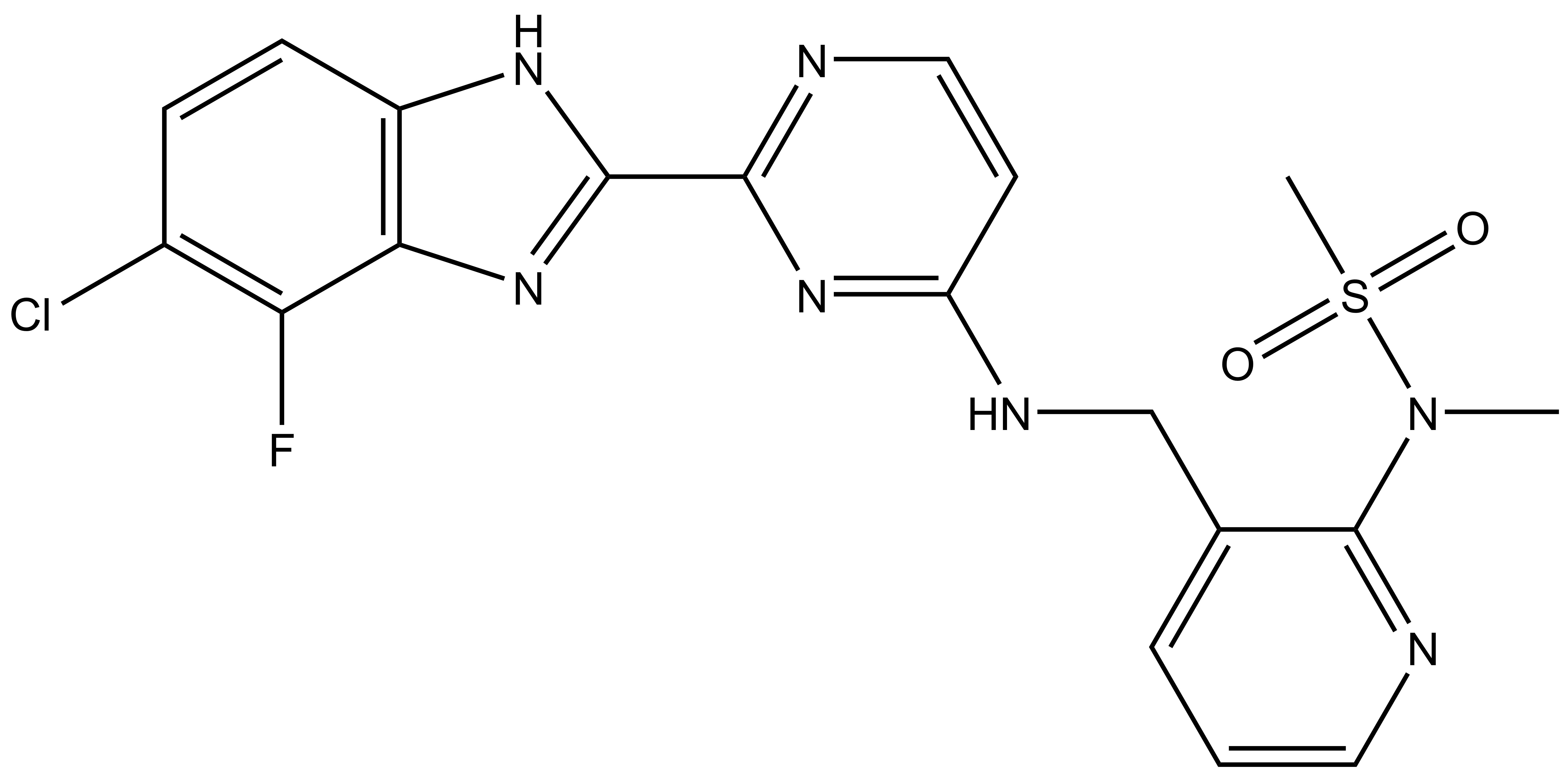
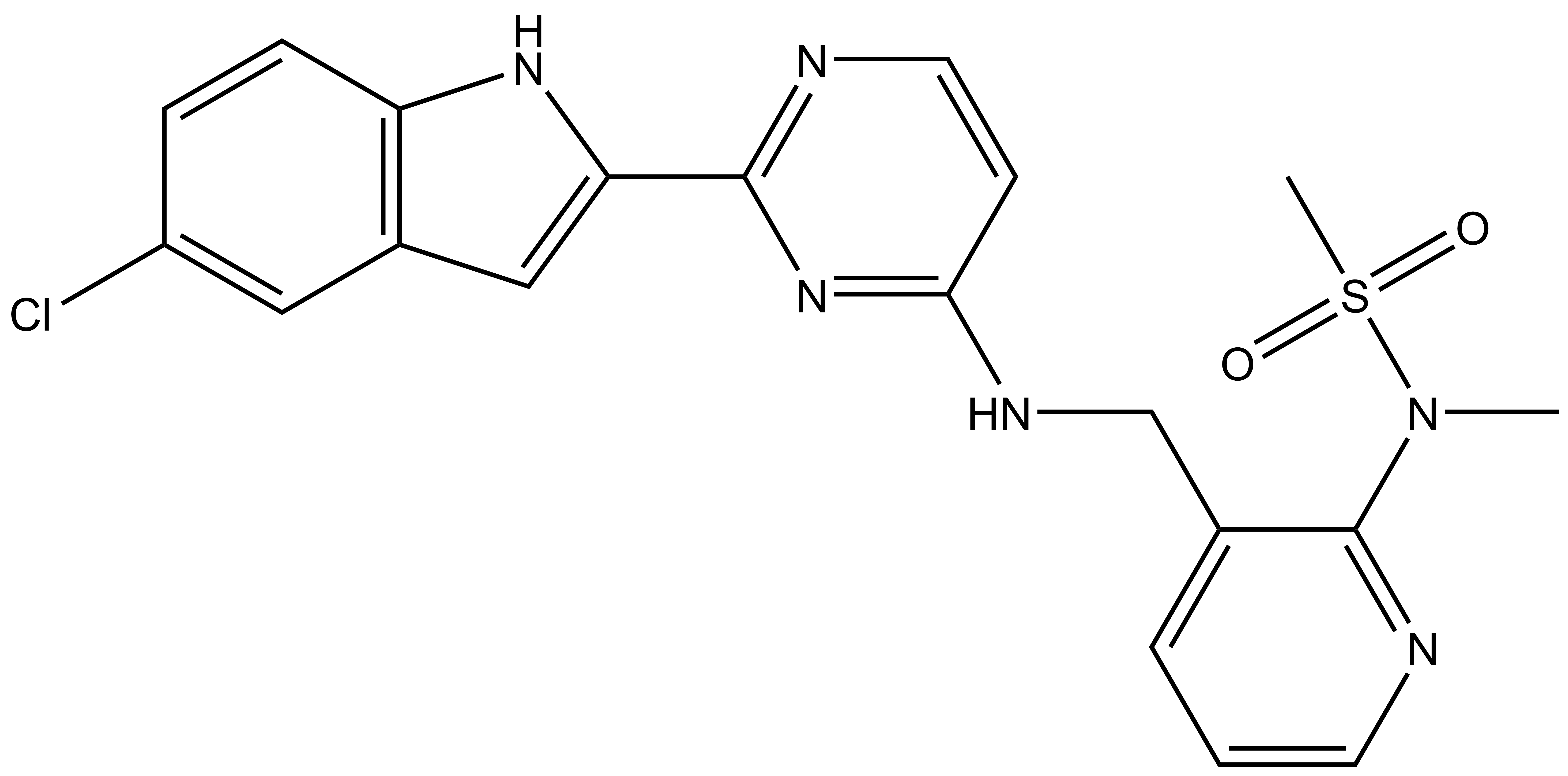
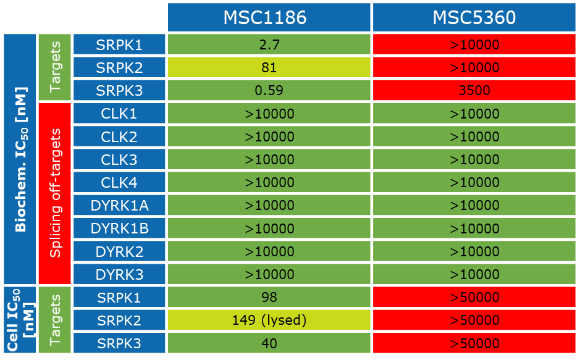
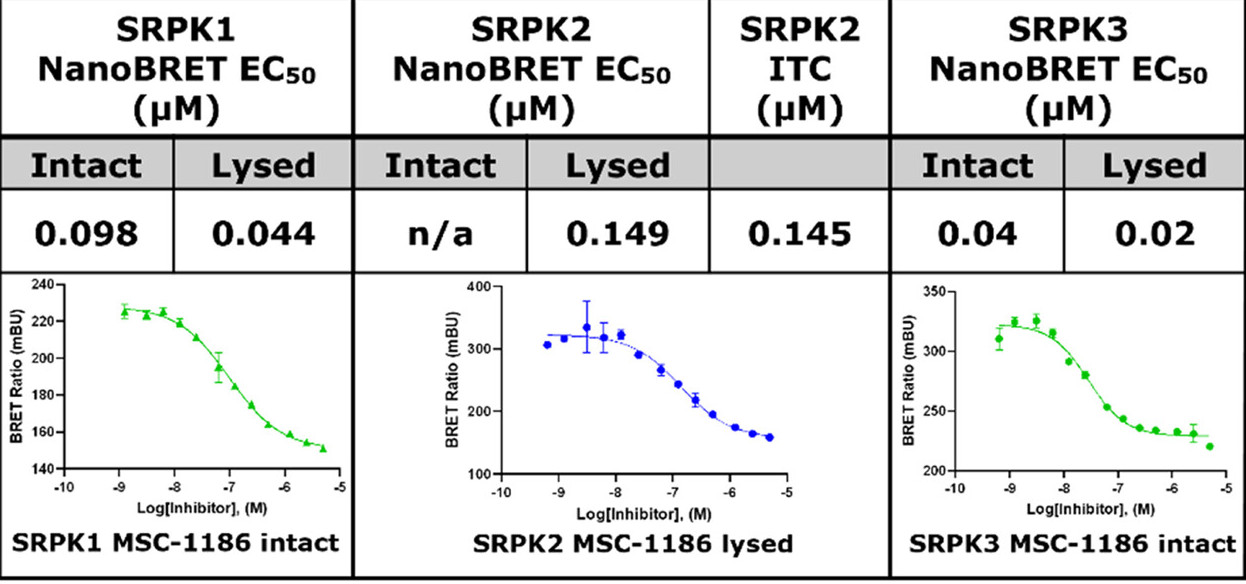
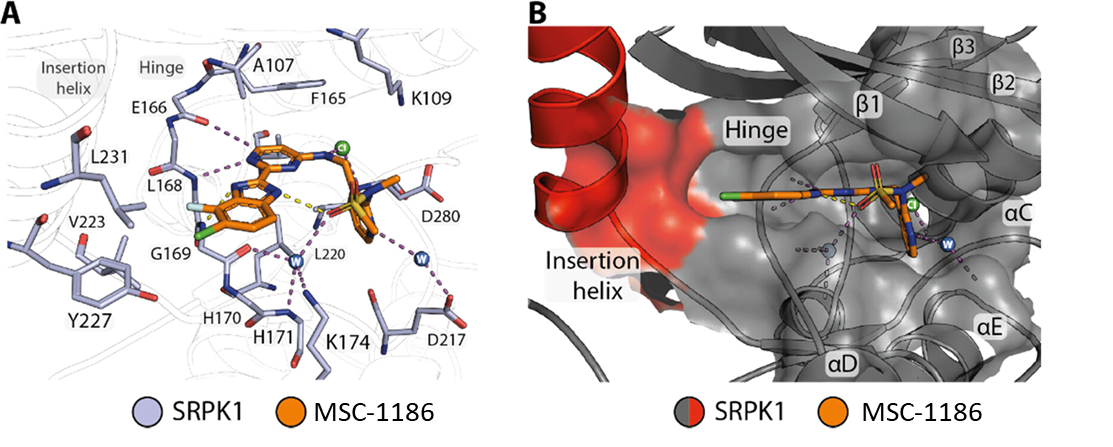
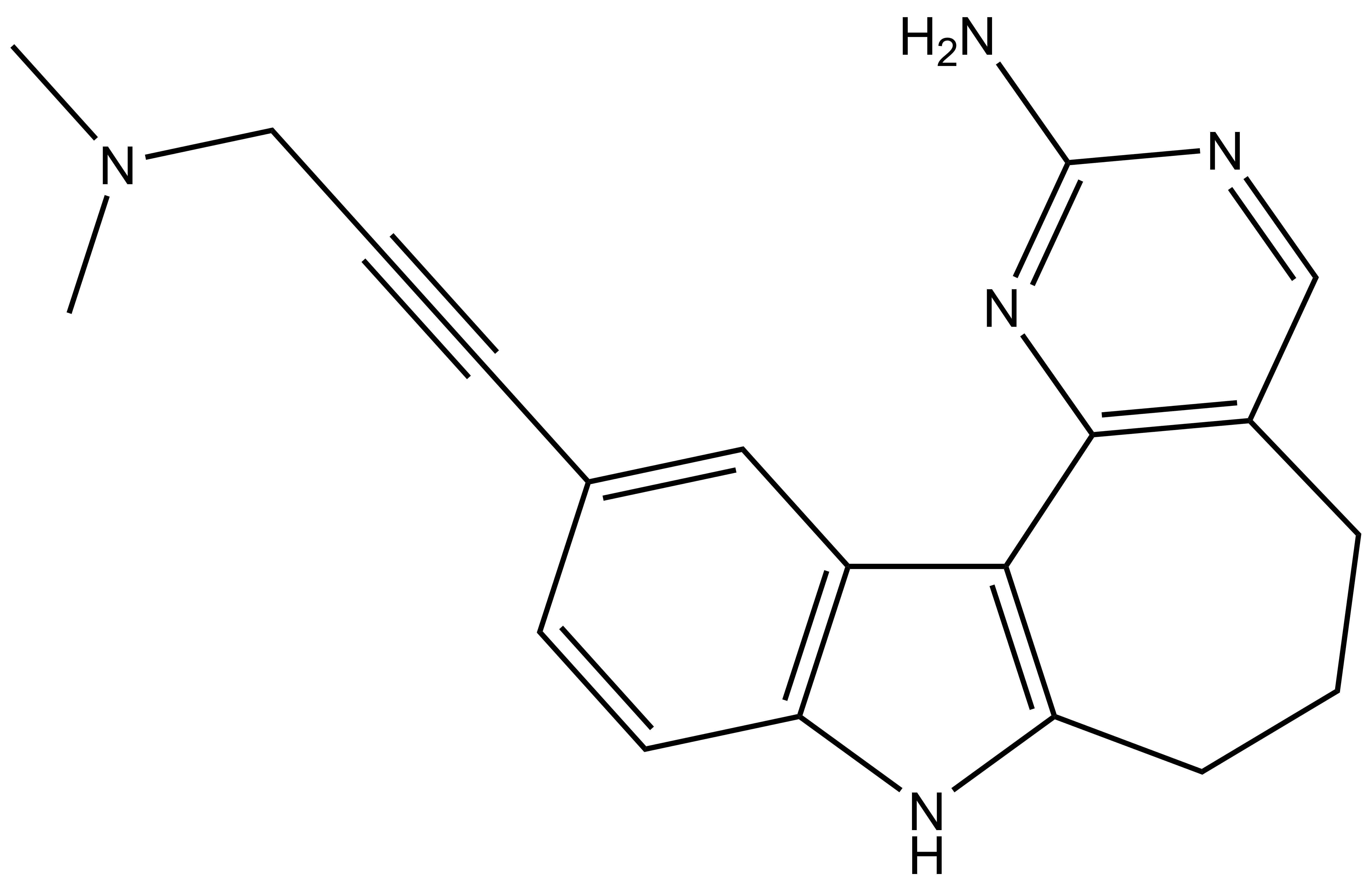
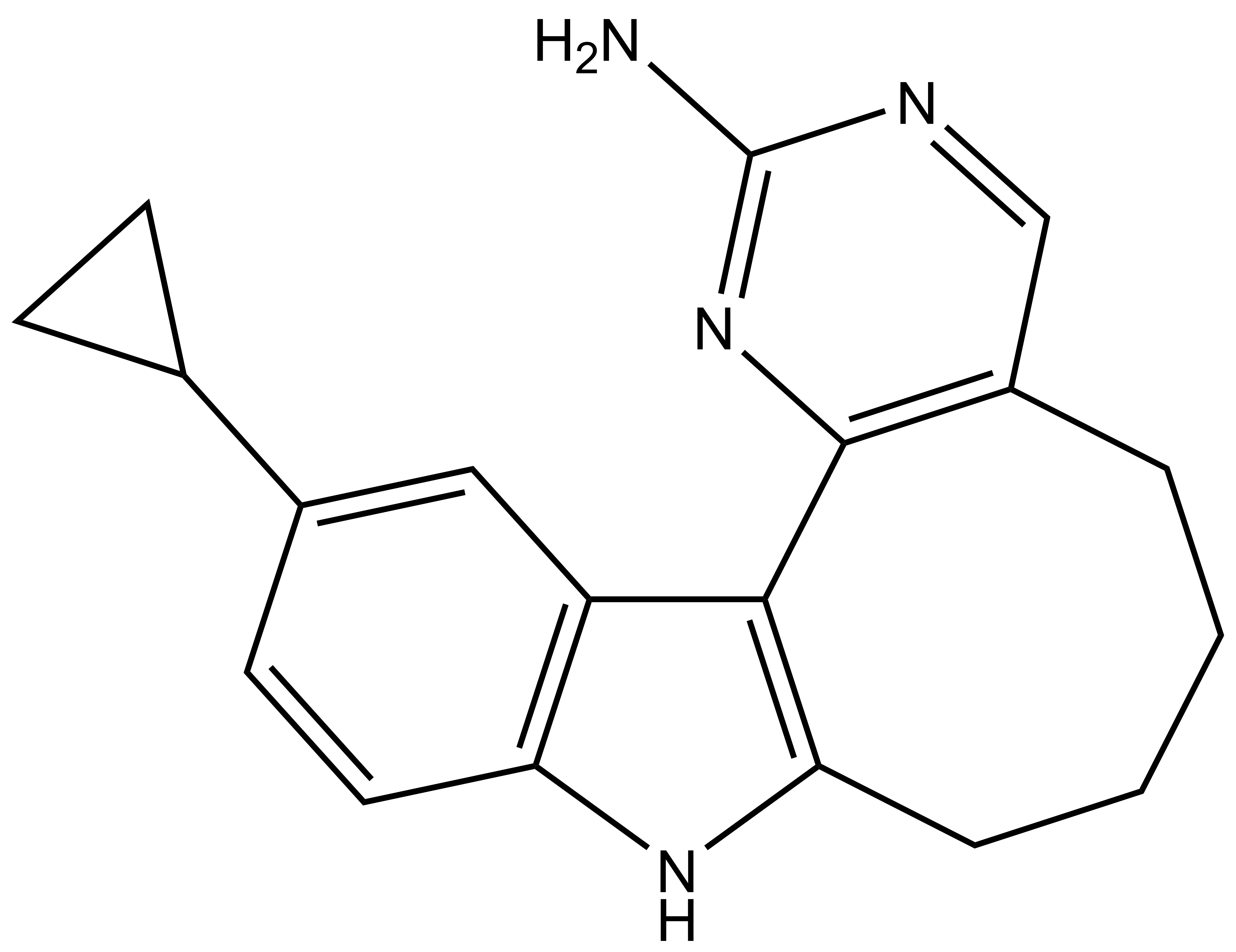
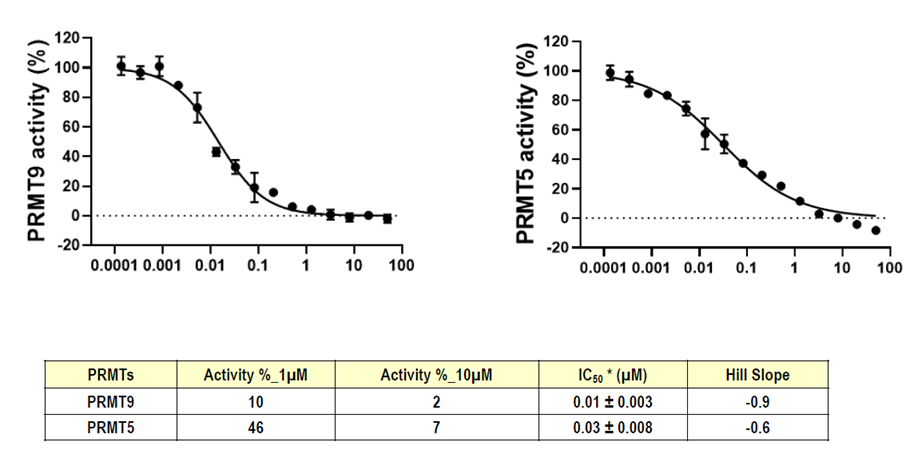
Takeda in collaboration with the SGC has developed LIJTF500025, a potent and selective inhibitor for the LIMK1/2. LIJTF500025 binding to LIMK1/2 was confirmed by DSF and ITC. Its crystall structure revealed an allosteric binding mode (Type III) that explains he high selectivity within the humane kinome. The cellular activity was determined by NanoBRET and revealed EC50 values of 82 nM on LIMK1 and 52 nM on LIMK2 was just one additional off-target (RIPK1 EC50 = 6 nM). The chemical probe (LIJTF500025) is accompanied by a negative control (LIJTF500120) that is structurally closely related to the probe molecule.
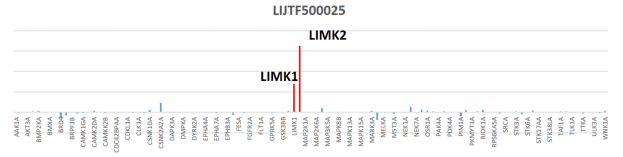
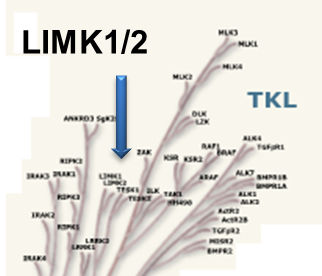
Potency Against Target Family
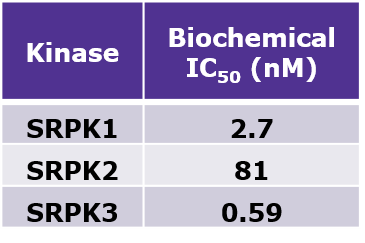
LIJTF500025 had an KD Value of 37 nM on LIMK1 determined by ITC.
Selectivity
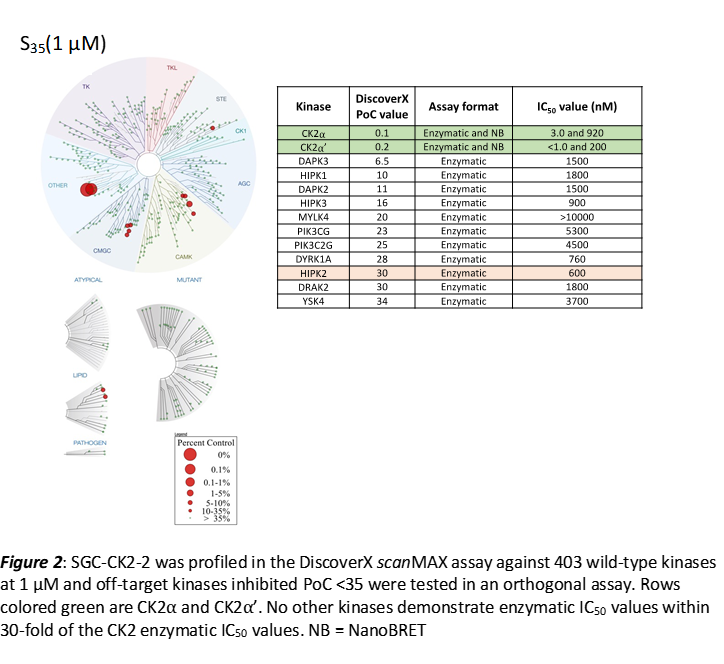
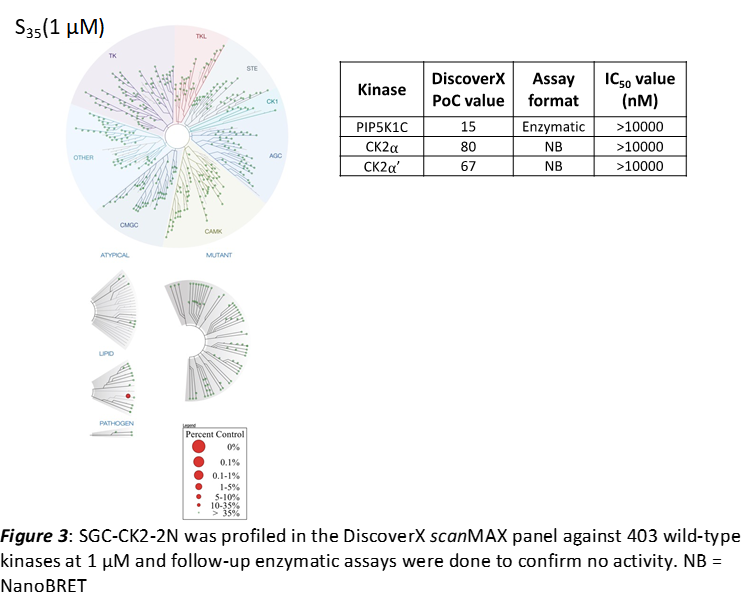
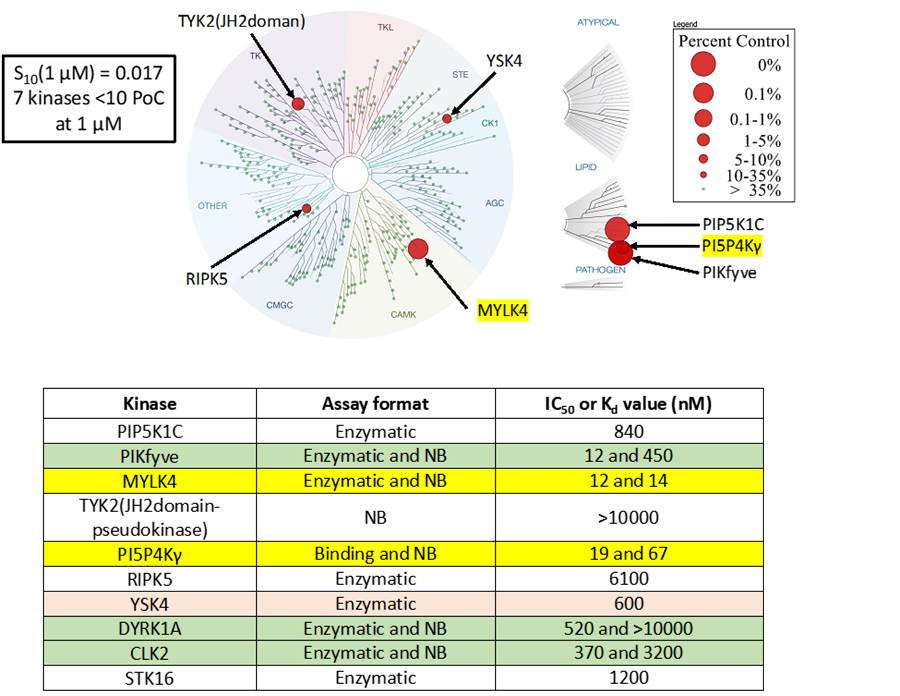
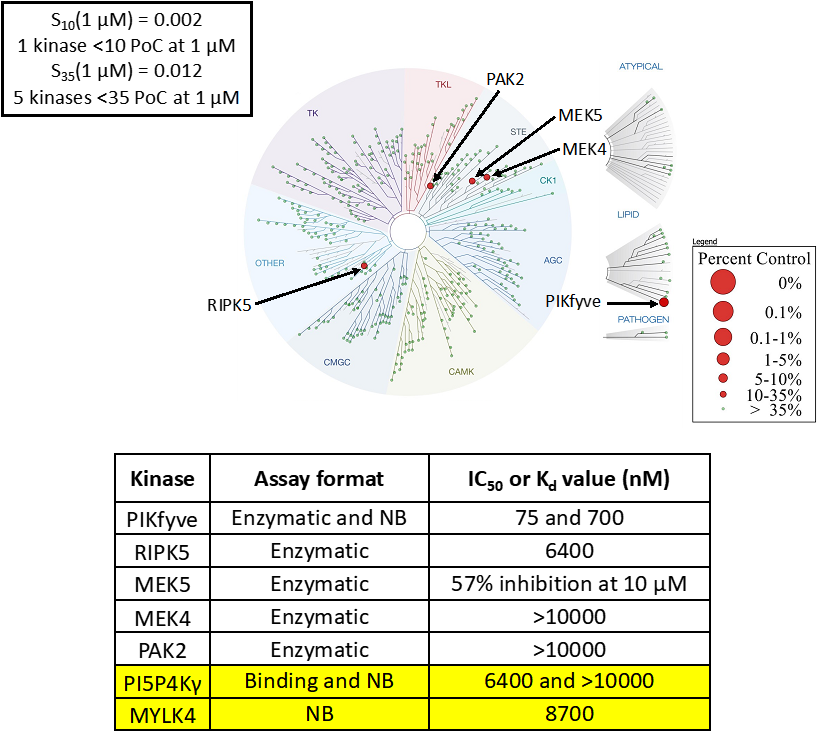
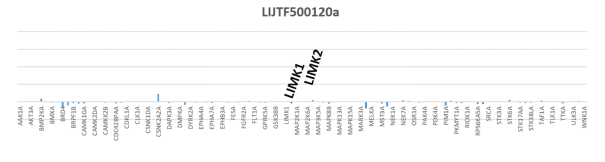
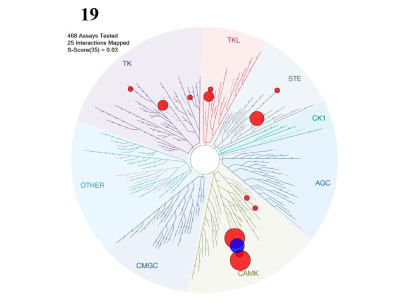
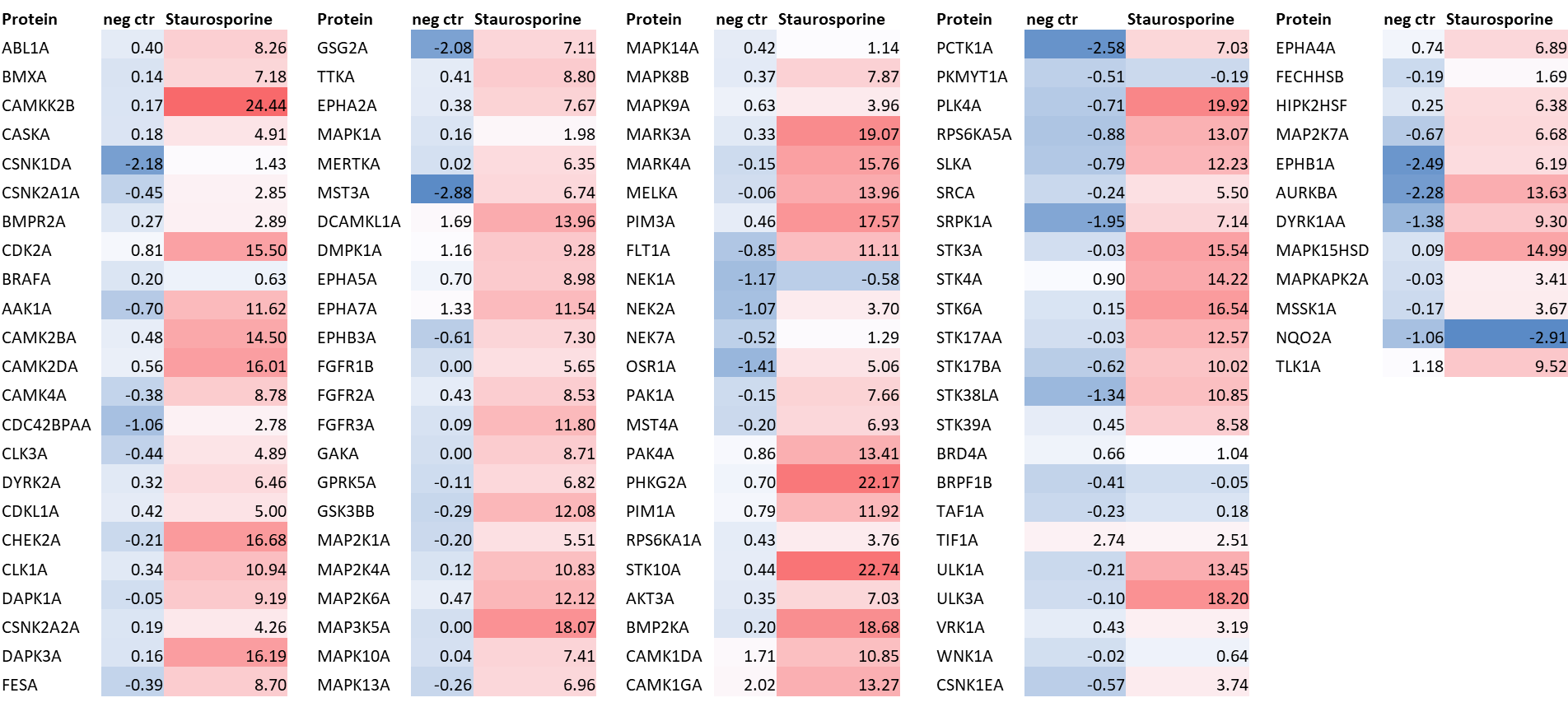
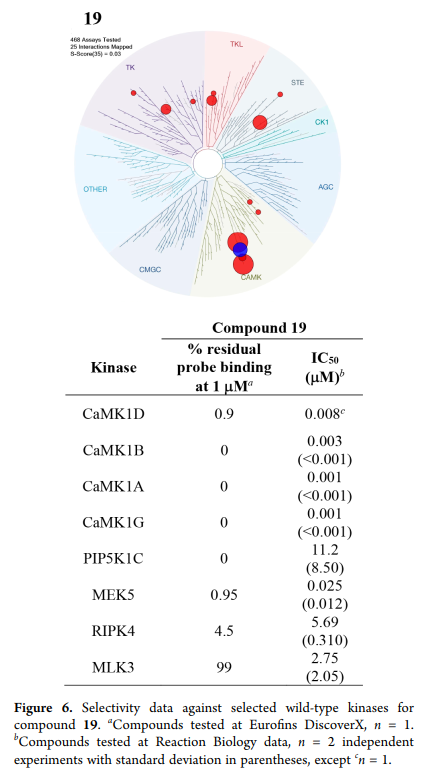
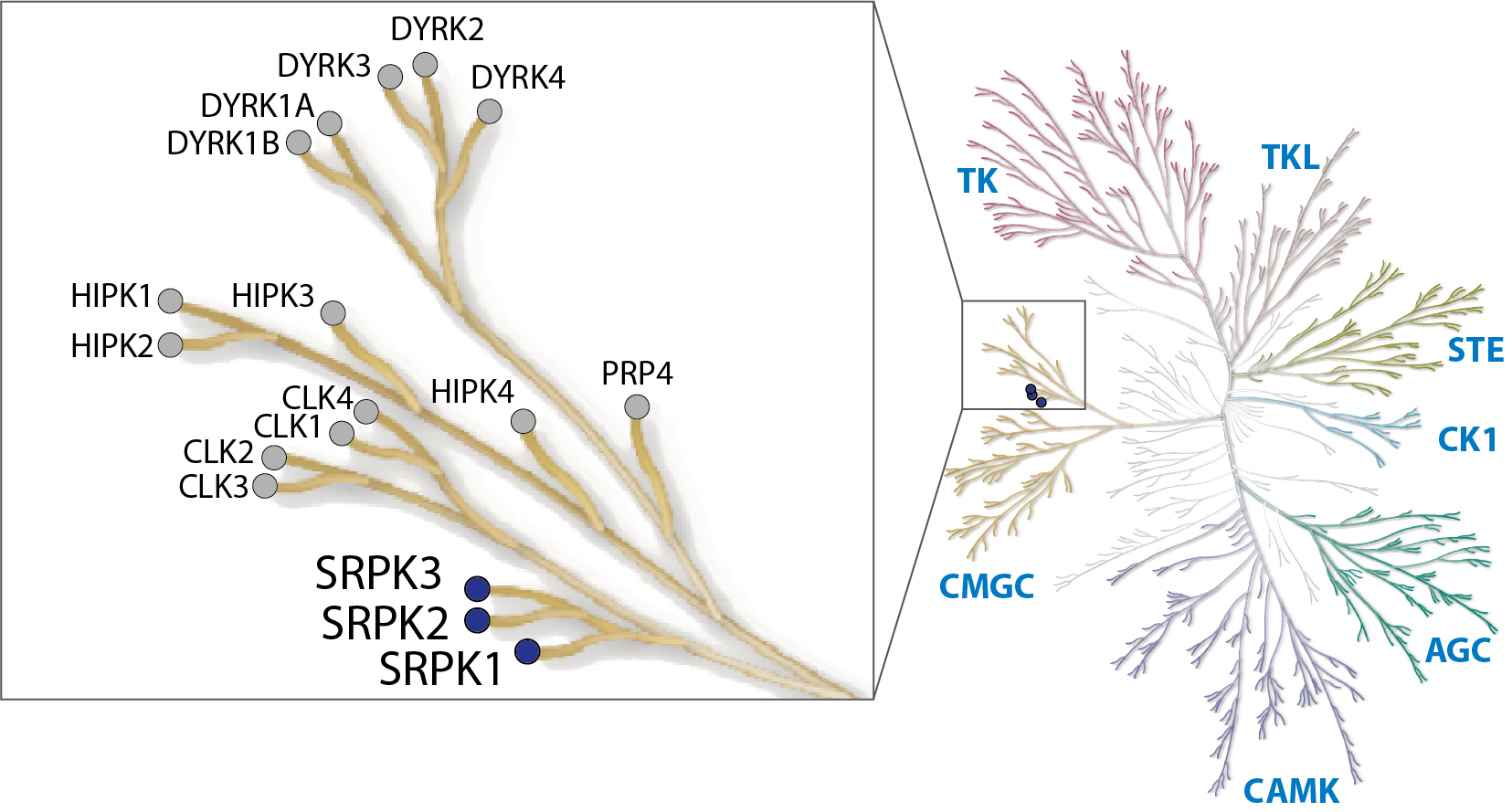
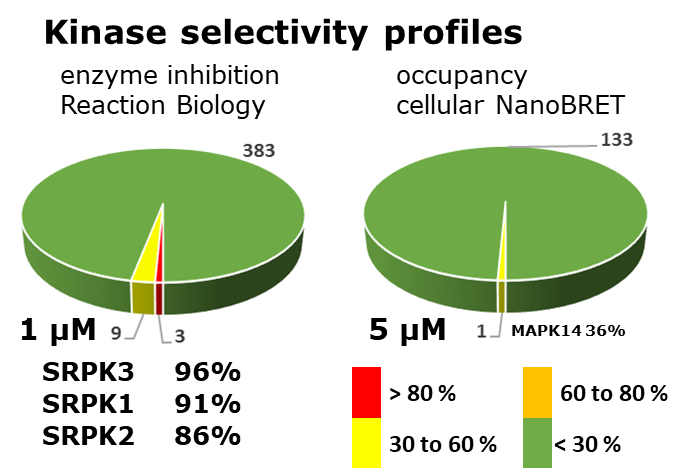
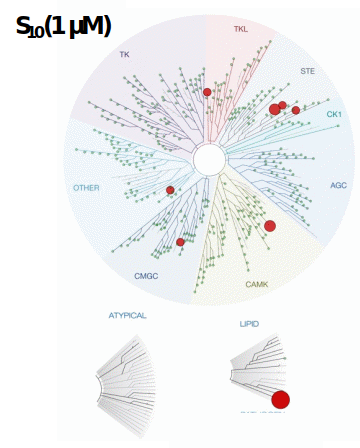
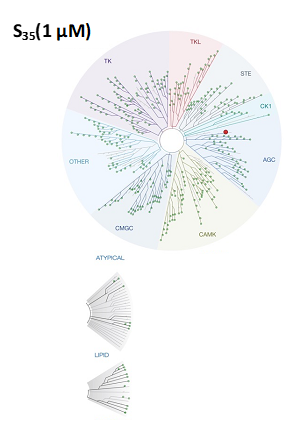
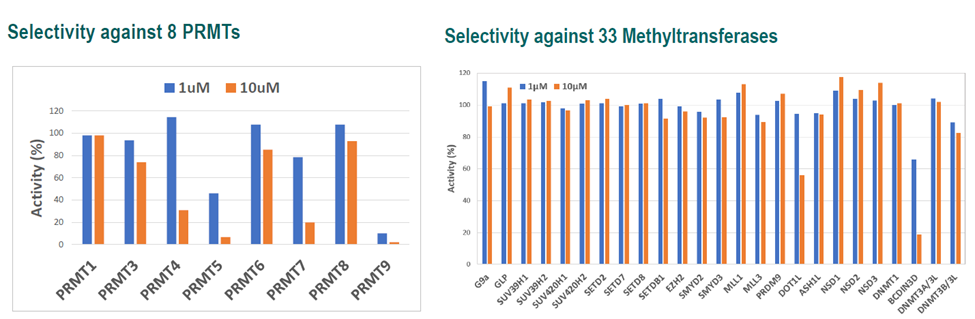
LIJTF500025 was selective in an in-house DSF kinase panel against 107 kinases. In addition, the selectivity was confirmed in the ScanMAX Kinase Panel from Eurofins (DiscoverY) against 468 kinases at a screening concentration of 1 µM.
Dosage
Based on the potency and the selectivity of the chemical probe and to minimize the risk of unspecific cytotoxicity, we recommend a concentration of no higher than 1 µM for cell-based assays.
Cellular Activity
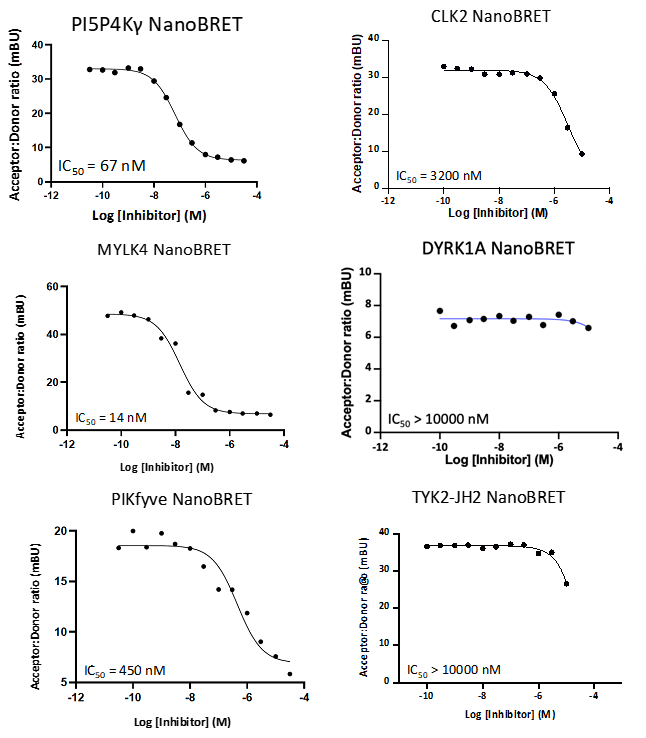
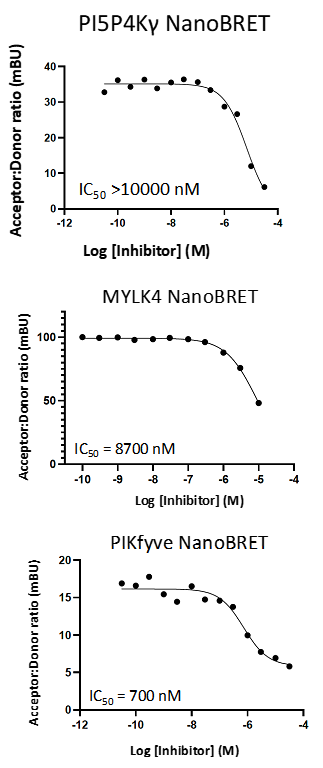
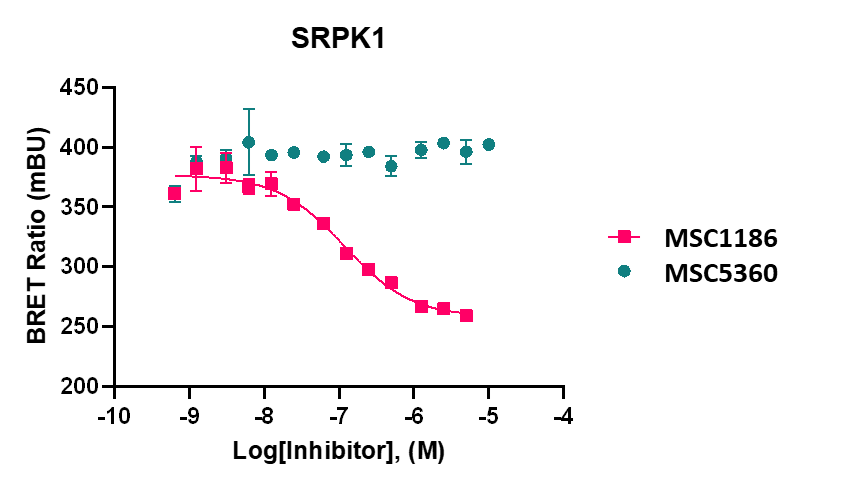
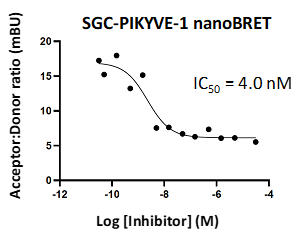
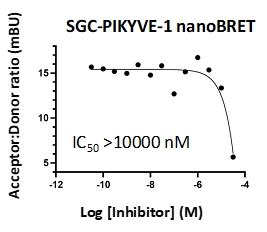
LIJTF500025 displayed an EC50 of 82 nM, 52 nM and 6.3 nM on LIMK1, LIMK2 and RIPK1 respectively in intact cells in the NanoBRET assay.