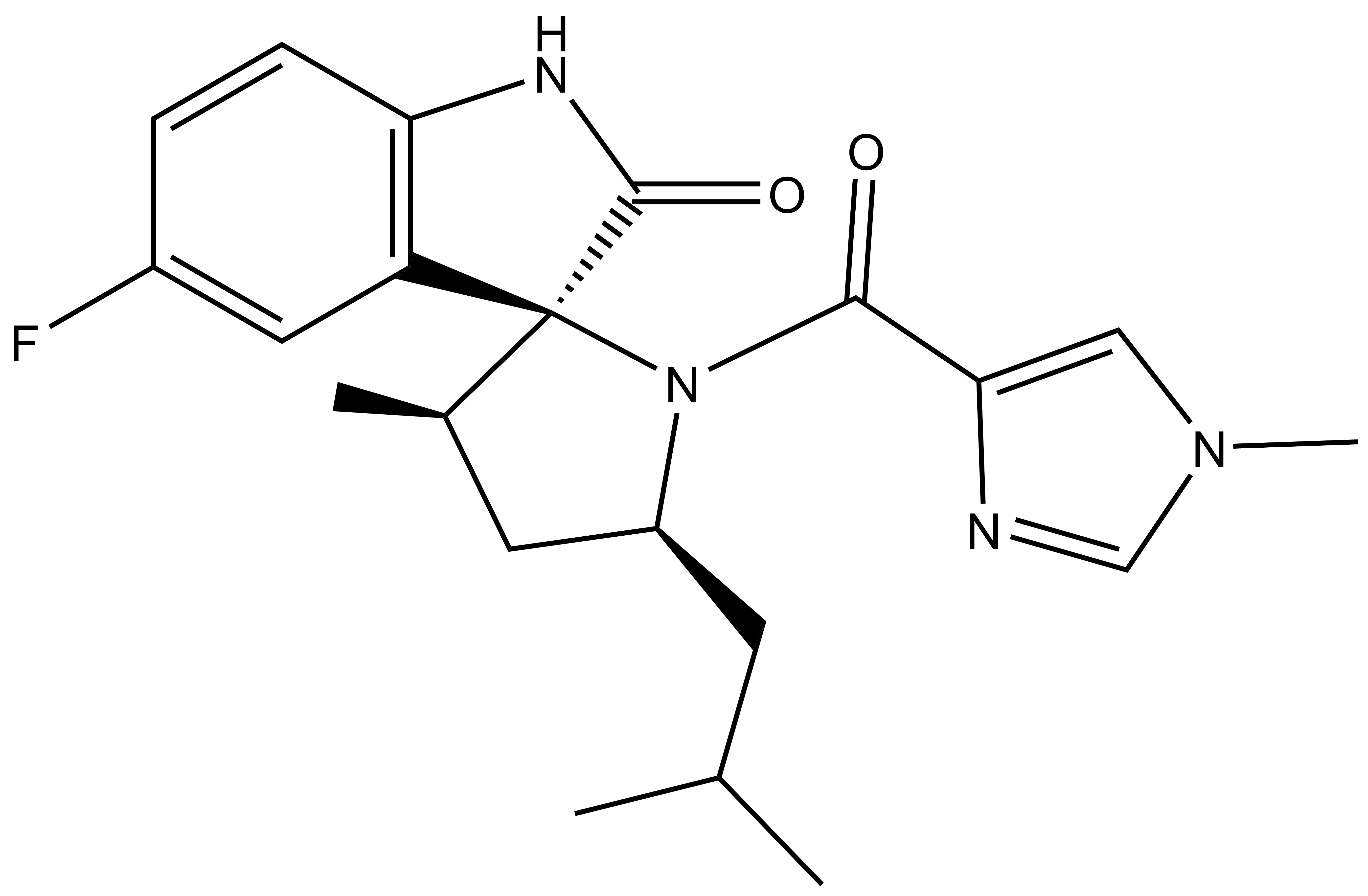
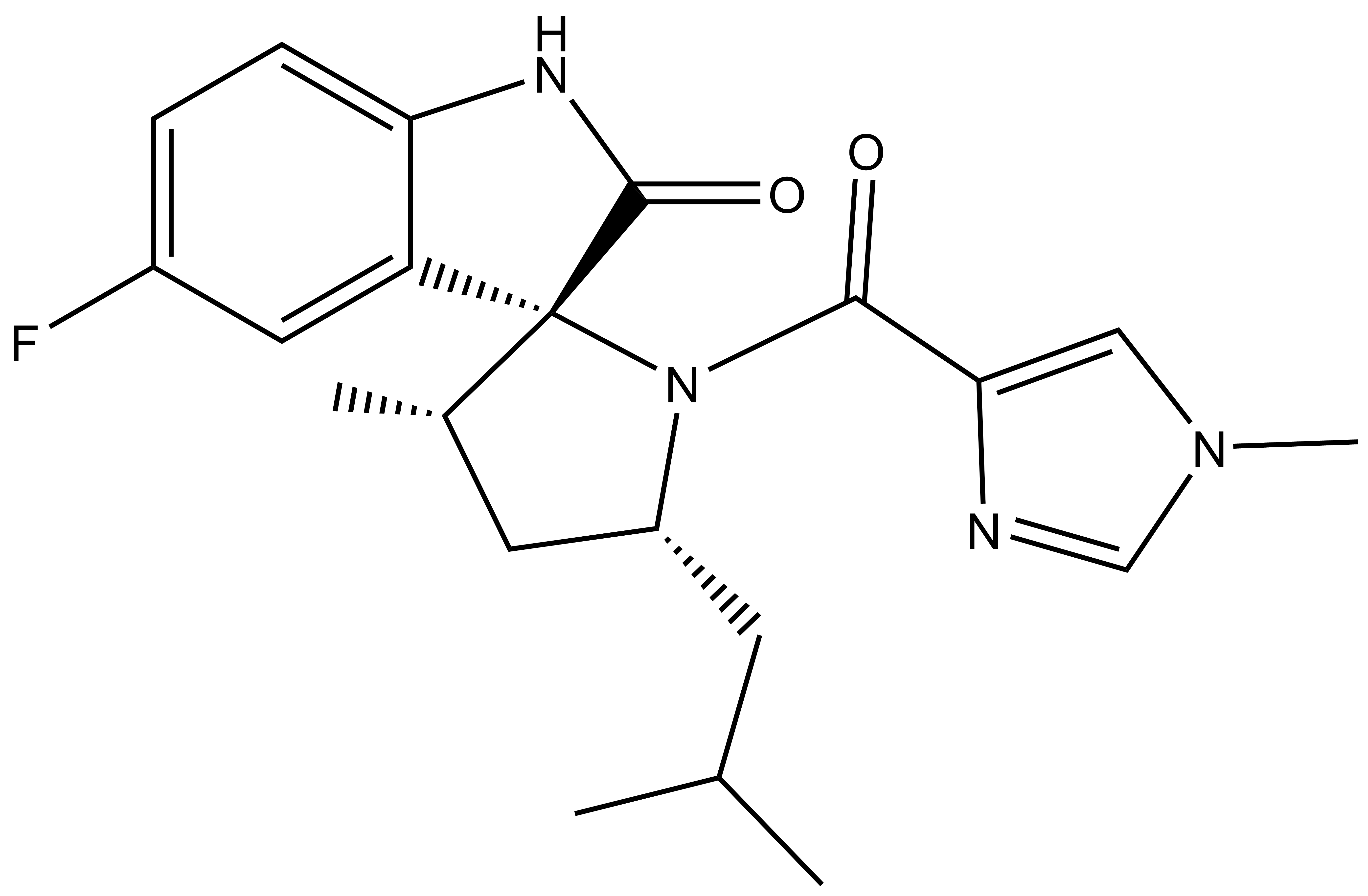
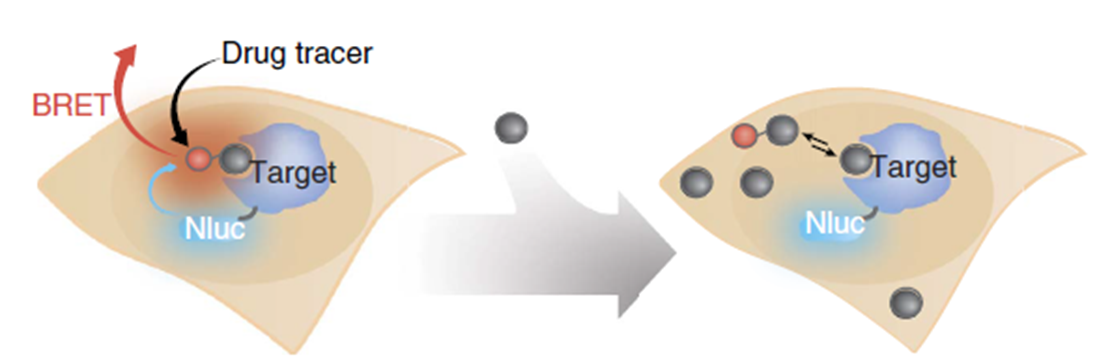
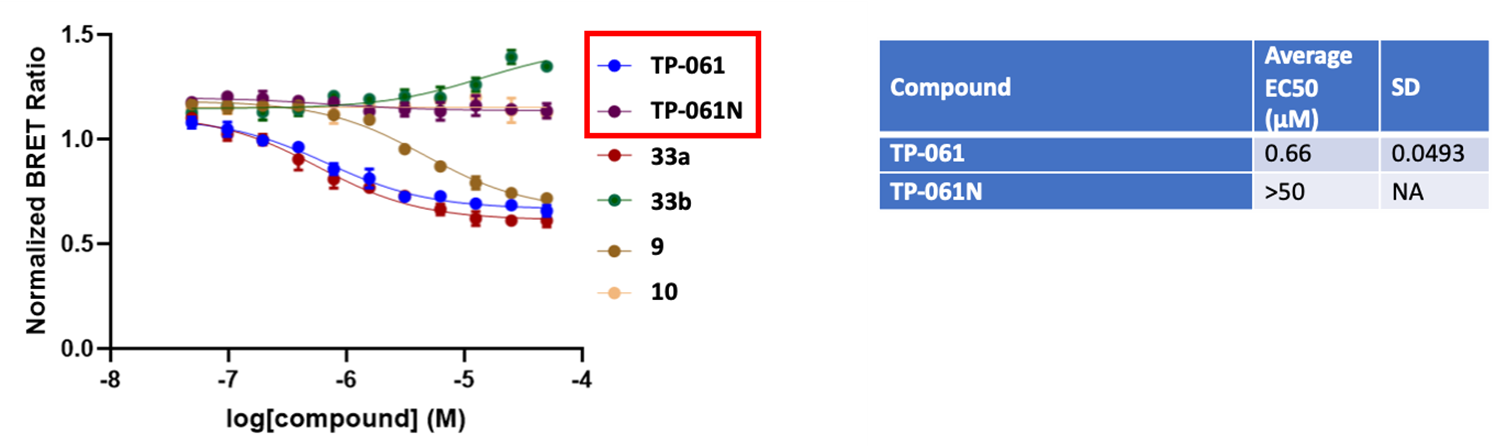
13.03.2024
Structural Genomics Consortium Principal Investigators Receive $4.3 Million Infrastructure Award to Transform Early Drug Discovery at the University of Toronto
Toronto, March 13, 2024 – The Structural Genomics Consortium’s (SGC) Principal Investigators at the University of Toronto have been awarded a $4.3-million infrastructure award from the Canada Foundation for Innovation (CFI) and the Ontario Research Fund (ORF) through the 2023 Innovation Fund competition.