Left to right: Marianne van Hage, Andreas Gidlöf, Mireia Cruz de los Santos, Claes Andersson, Alexandra Argyriou, Pia Hydén, Johan Weigelt (Photo courtesy of Michael Sundström).
Cheryl Arrowsmith, Chief Scientist of the Structural Genomics Consortium (SGC), Senior Scientist at the Princess Margaret Cancer Centre, and Professor at the Department of Medical Biophysics at the University of Toronto, has been honoured by Clarivate as one of the Highly Cited Researchers for 2023.
The probe and control may be requested here.
| Probe | Negative control | |
 |
|  |
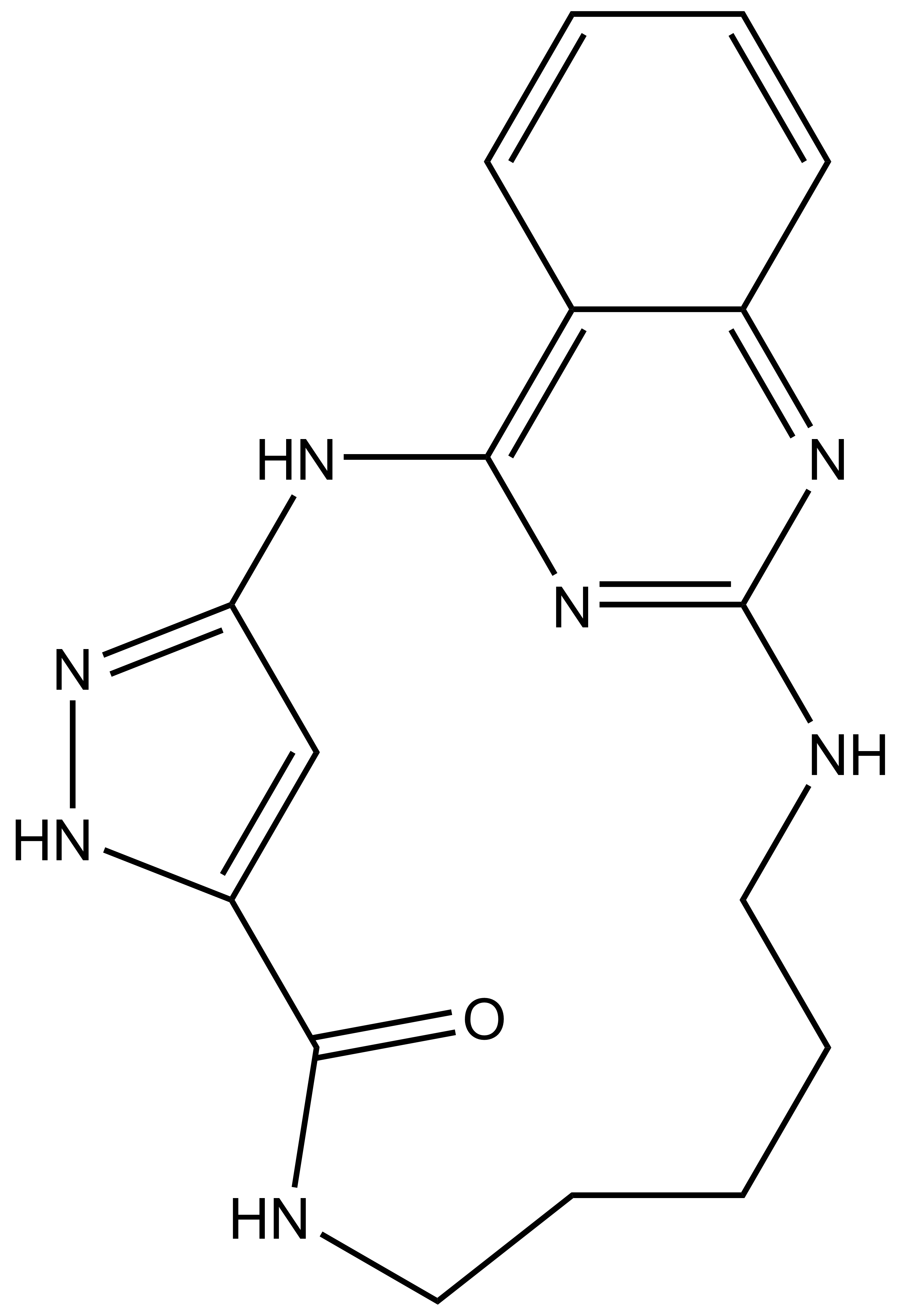
JA310 |
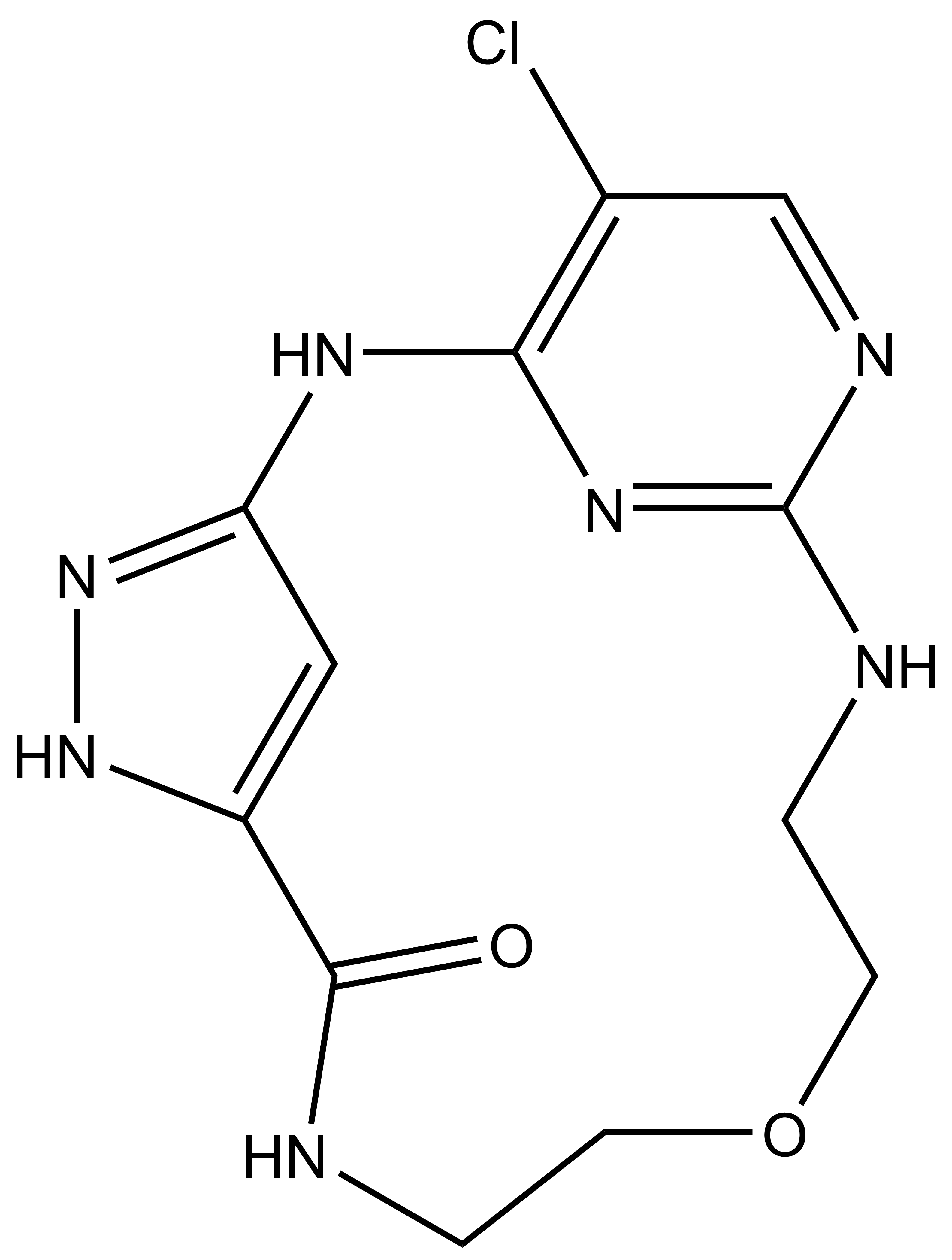
| JA262 |
Mammalian STE20-like protein kinase 3 and -4 (MST3/4) are part of the MST family, which comprises three additional members, namely MST1, MST2 and YSK1. The MSTs are involved in cell proliferation, cell migration and cell polarity.[1] For MST3 it has been shown that it can phosphorylate STK38L and stimulate its kinase activity [2]; for MST4 ATG4B has been demonstrated as a substrate and thereby regulates the autophagic activity.[3]
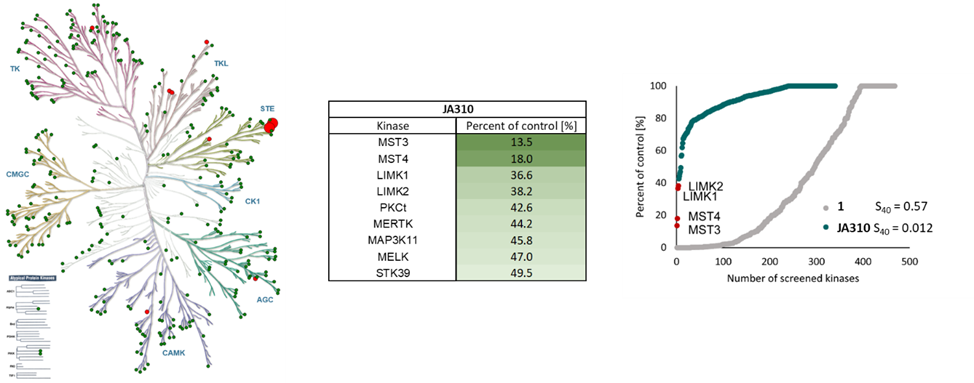
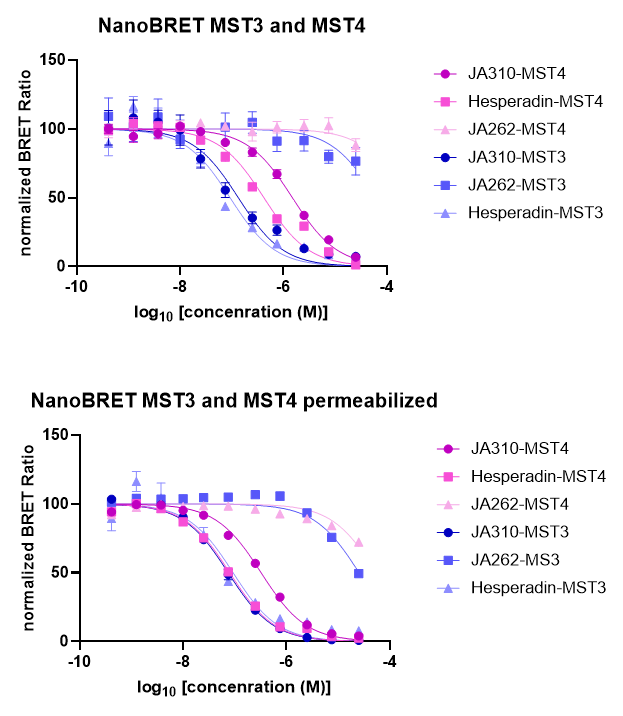
SGC has developed JA310, a potent and selective MST3 inhibitor with a EC50 determined by a NanoBret of 106 nM for MST3 in intact cells and 76 nM in permeabilized cells. JA310 shows kinome wide selectivity in a screening against 340 WT kinases (from Reaction Biology). In addition, the chemical probe (JA310) is accompanied by a negative control (JA262), which is structurally closely related to the probe molecule.[4]
Selectivity
JA310 has been shown to be selective in an in vitro kinase panel from Reaction Biology against 340 WT-Kinases followed by cellular NanoBRET assays.
Dosage
To distinguish the effect between MST3 and MST4 in a cell-based assays, we recommend to use JA310 together with the neg. control compound JA262 at a concentration of no higher than 1 µM for cell-based assays.
Cellular Activity
JA310 displayed an EC50 of 106 nM in the NanoBRETTM assay.
| Probe | Negative control | |
|
| |
JA310 |
| JA262 |
Physical and chemical properties JA310 | |
| MW | 337.39 |
| MF | C17H19N7O |
| IUPAC name | 2,4,5,8,14,16,23-heptaazatetracyclo[13.7.1.1³,⁶.0¹⁷,²²]tetracosa-1(22),3,6(24),15(23),16,18,20-heptaen-7-one |
| logP | 2.46 |
| PSA | 107.62 |
| No. of chiral centres | 0 |
| No. of rotatable bonds | 0 |
| No. of hydrogen bond acceptors | 6 |
| No. of hydrogen bond donors | 4 |
| Storage | stable as solid in the dark at -20°C. NB making aliquots rather than freeze-thawing is recommended |
| Dissolution | soluble in DMSO in a concentration of 10 mM |
SMILES: O=C(C1=CC(N2)=NN1)NCCCCCNC3=NC2=C4C=CC=CC4=N3
InChI: InChI=1S/C17H19N7O/c25-16-13-10-14(24-23-13)21-15-11-6-2-3-7-12(11)20-17(22-15)19-9-5-1-4-8-18-16/h2-3,6-7,10H,1,4-5,8-9H2,(H,18,25)(H3,19,20,21,22,23,24)
InChIKey: XBWKVZHZUZNMJE-UHFFFAOYSA-N
Physical and chemical properties JA262 | |
| MW | 323.74 |
| MF | C12H14ClN7O2 |
| IUPAC name | 18-chloro-11-oxa-2,4,5,8,14,16,19-heptaazatricyclo[13.3.1.1³,⁶]icosa-1(18),3,6(20),15(19),16-pentaen-7-one |
| logP | 0.46 |
| PSA | 116.85 |
| No. of chiral centres | 0 |
| No. of rotatable bonds | 0 |
| No. of hydrogen bond acceptors | 7 |
| No. of hydrogen bond donors | 4 |
| Storage | stable as solid in the dark at -20°C. NB making aliquots rather than freeze-thawing is recommended |
| Dissolution | soluble in DMSO in a concentration of 10 mM |
SMILES: O=C(C1=CC(N2)=NN1)NCCOCCNC3=NC2=C(C=N3)Cl
InChI: InChI=1S/C12H14ClN7O2/c13-7-6-16-12-15-2-4-22-3-1-14-11(21)8-5-9(20-19-8)17-10(7)18-12/h5-6H,1-4H2,(H,14,21)(H3,15,16,17,18,19,20)
InChIKey: SXVMAWWNAHHYKH-UHFFFAOYSA-N
Selectivity profile of JA310 was determined with the selectivity profiling assay against 340 wild-type protein kinases from Reaction Biology at 1000 nM and on- and off targets were evaluated in cellulo EC50 with NanoBRET assay.
Kinase | Percent of control(%) @ 1000 nM | NanoBRET EC50 (µM) |
| MST3 | 13.5 | 0.11 |
| MST4 | 18.0 | 1.43 |
| LIMK1 | 36.6 | 5.67 |
| LIMK2 | 38.2 | 1.78 |
| PKCt | 42.6 | > 50.0 |
| MERTK | 44.2 | 5.14 |
| MAP3K11 | 45.8 | > 50.0 |
| MELK | 47.0 | 45.7 |
| STK39 | 49.5 | n.d. |
The negative control JA262 showed no activity on a DSF assay for 102 kinases and no on-target activity determined by NanoBRET in intact cells and in lysed cells.
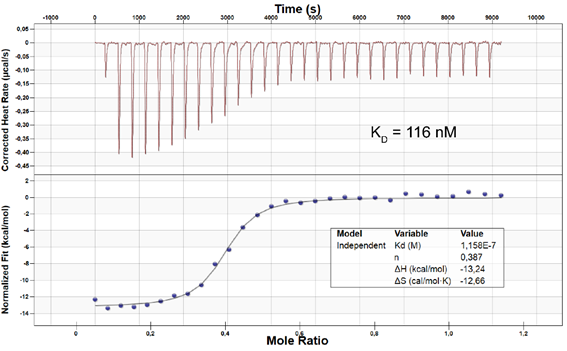
ITC on MST4: KD = 116 nM
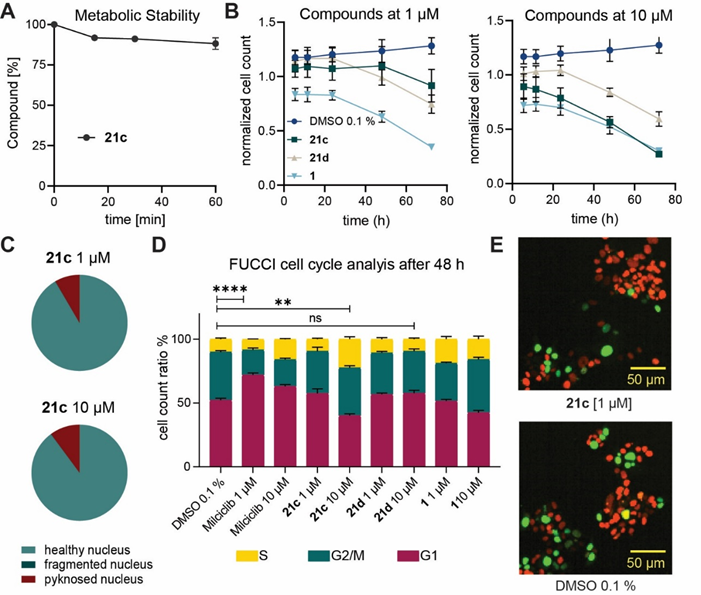
JA310 (21c) is metabolic stable. JA310 (21c) shows no general cytotoxicity after 24h, but after 72h a decrease of the normalized cell count has been detected. At 1 µM JA310 (21c) shows slightly increase of cells in G1 Phase, whereas at higher concentration a S-Phase arrest has been observed.
[1] Thompson BJ, Sahai E. MST kinases in development and disease. J Cell Biol. 2015 Sep 14;210(6):871-82. doi: 10.1083/jcb.201507005.
[2] Cornils H, Kohler RS, Hergovich A, Hemmings BA. Human NDR kinases control G(1)/S cell cycle transition by directly regulating p21 stability. Mol Cell Biol. 2011 Apr;31(7):1382-95. doi: 10.1128/MCB.01216-10.
[3] Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X, Shi T, Yang Y, Sastry N, Horbinski CM, Lu S, Stupp R, Kessler JA, Nishikawa R, Nakano I, Sulman EP, Lu X, James CD, Yin XM, Hu B, Cheng SY. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell. 2017 Dec 11;32(6):840-855.e8. doi: 10.1016/j.ccell.2017.11.005.
[4] Jennifer A. Amrhein, Lena M. Berger, Dimitrios-Ilias Balourdas, Andreas C. Joerger, Amelie Menge, Andreas Krämer, Julia M. Frischkorn, Benedict-Tilman Berger, Lewis Elson, Astrid Kaiser, Manfred Schubert-Zsilavecz, Susanne Müller, Stefan Knapp, Thomas Hanke. Synthesis of pyrazole-based macrocycles leads to a highly selective inhibitor for MST3. bioRxiv 2023.10.20.563248; doi: https://doi.org/10.1101/2023.10.20.563248
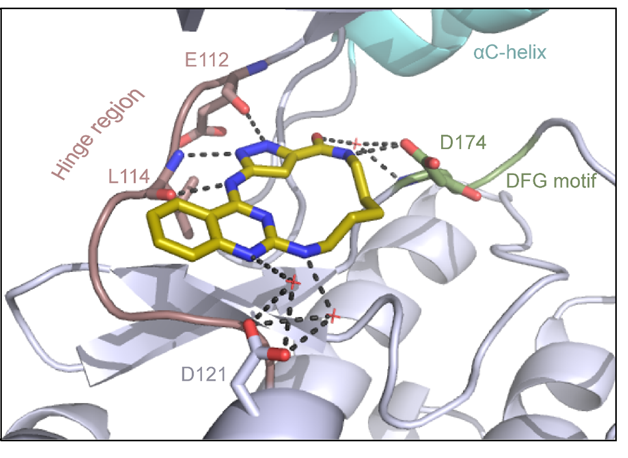
Binding mode of JA310 in complex with MST3 was determined (PDB 8QLQ). The inhibitor binds in an orthosteric binding mode to the ATP pocket as a type I kinase inhibitor.
Government General Mary Simon has announced 53 new Order of Canada appointments- one of Canada’s highest honours. This prestigious recognition celebrates individuals from diverse backgrounds for their outstanding contributions to Canada.
Established in 1067, the Order of Canada acknowledges remarkable achievements, dedication to the community and service to the nation, with over 7,600 people spanning various sectors invested in making a difference in Canada.
TORONTO, Canada, October 3, 2023 – The Structural Genomics Consortium (SGC) today announced that it has entered into a partnership with Shanghai Stock Exchange listed company HitGen Inc. ("HitGen", SSE: 688222.SH). HitGen will utilize its DNA-encoded library (DEL) technology platform, specifically OpenDEL™, to screen under-represented targets chosen by SGC.