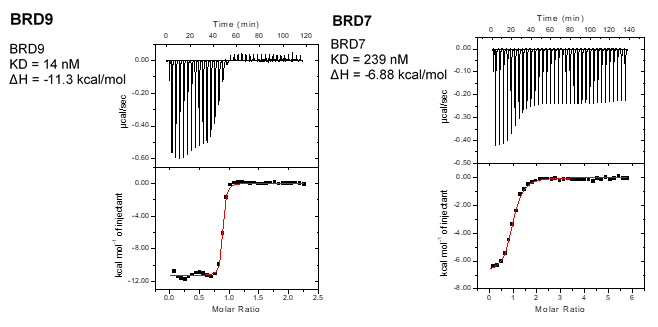
Isothermal Titration Calorimetry (ITC)
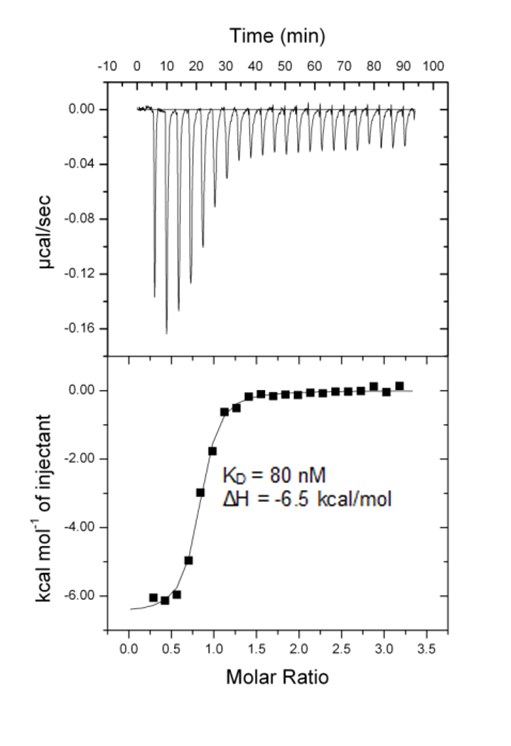
All calorimetric experiments were performed on a VP-ITC micro-calorimeter (MicroCalTM, LLC Northampton, MA). Protein solutions were buffer exchanged by gel filtration or dialysis into buffer (20 mM Hepes pH 7.5, 150 mM NaCl, and 0.5 mM tris (2-carboxyethyl) phosphine (TCEP). All measurements were carried out at 288.15 K. All injections were performed using an initial injection of 2 µL followed by injections of 8 µL. The data were analysed with the MicroCal ORIGIN software package employing a single binding site model. The first data point was excluded from the analysis.
Temperature shift assay
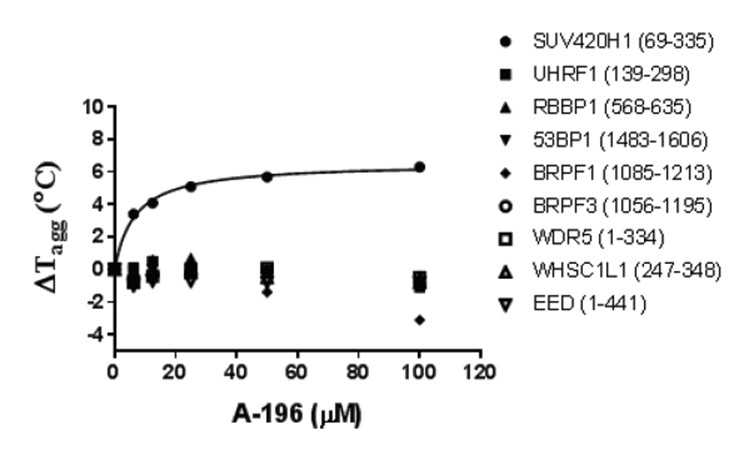
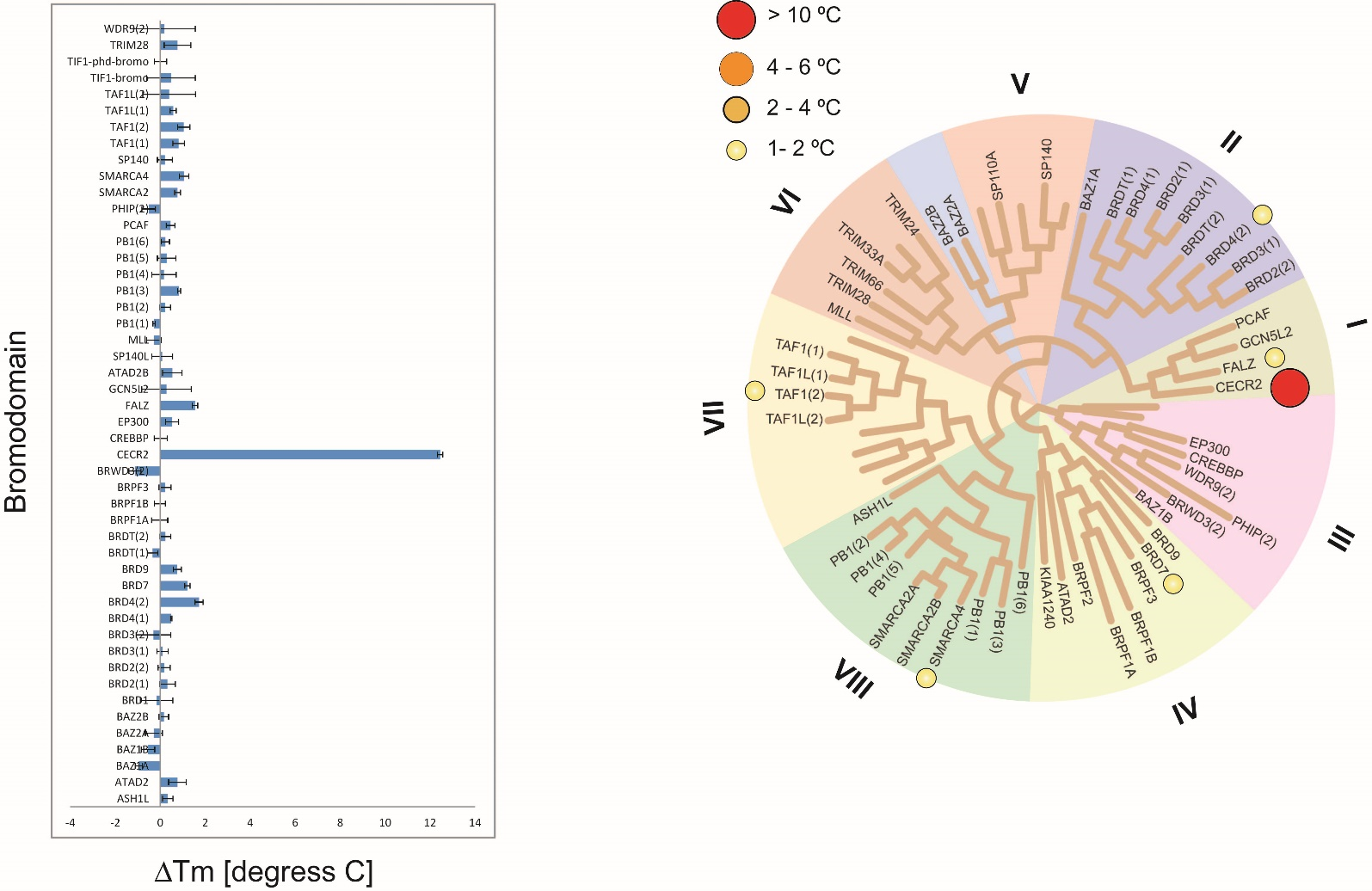
Thermal melting experiments were carried out using a Stratagene Mx3005p Real Time PCR machine (Agilent Technologies). NVS-CECR2-1 was added at a final concentration of 10 µM. SYPRO Orange (Molecular Probes) was added as a fluorescence probe at a dilution of 1:1000 as described (6).
AlphaScreen Assay
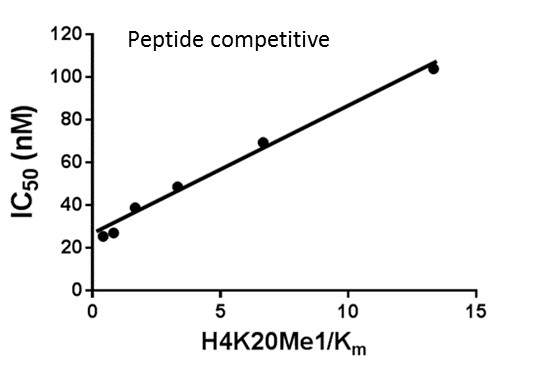
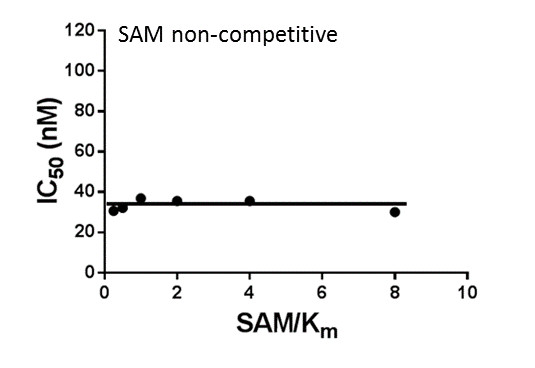
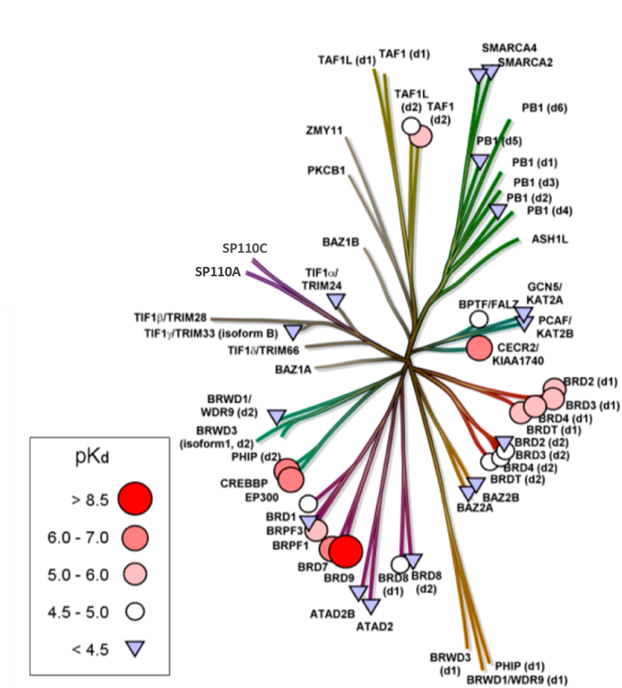
Assays were performed as described previously with minor modifications (7). All reagents were diluted in 25 mM HEPES, 100 mM NaCl, 0.1 % BSA, pH 7.4 supplemented with 0.05 % CHAPS and allowed to equilibrate to room temperature prior to addition to plates. An 11-point 1:2.0 serial dilutions of the ligands was prepared on lowvolume 384-well plates (ProxiPlateTM-384 Plus, PerkinElmer, USA), using LabCyte Eco liquid handler. Plates filled with 5 µL of the assay buffer followed by 7 µL of peptide H4K5acK8acK12acK16ac. Plates were sealed and incubated for a further 30 minutes, before the addition of 8 µM of the mixture of streptavidin-coated donor beads (12.5 µg/mL) and nickel chelate acceptor beads (12.5 µg/mL) under low light conditions. Plates were foil-sealed to protect from light, incubated at room temperature for 60 minutes and read on a PHERAstar FS plate reader (BMG Labtech, Germany) using an AlphaScreen 680 excitation/570 emission filter set. IC50 values were calculated in Prism 5 (GraphPad Software, USA) after normalization against corresponding DMSO controls.
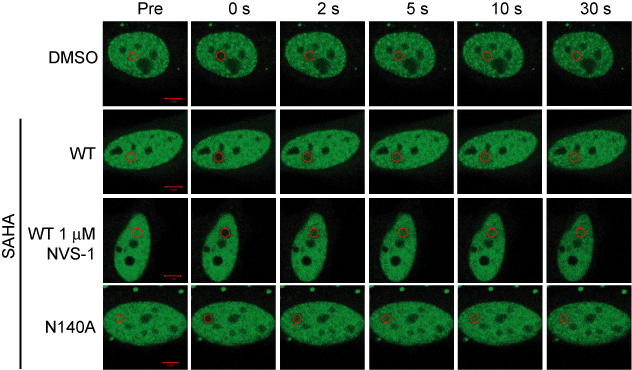
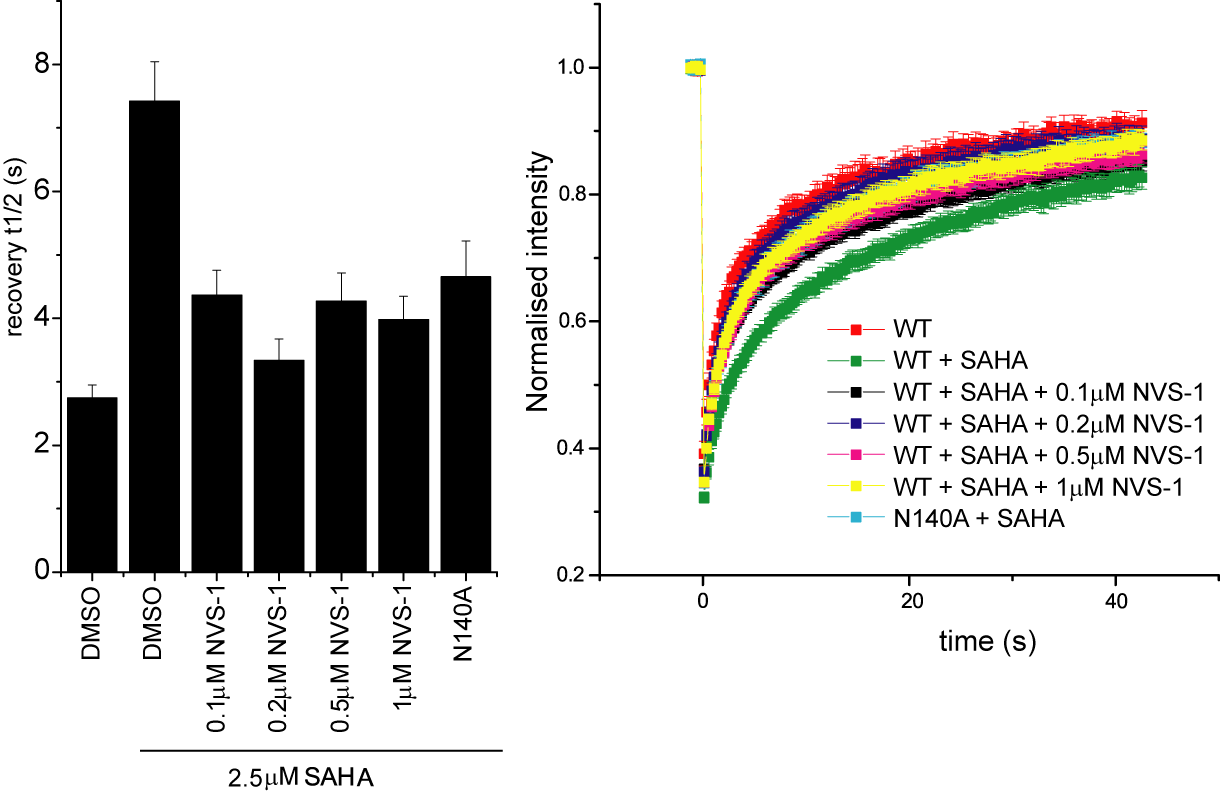
Fluorescence Recovery After Photobleaching (FRAP) Assay
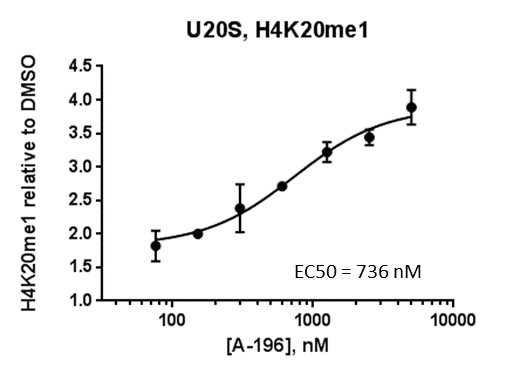
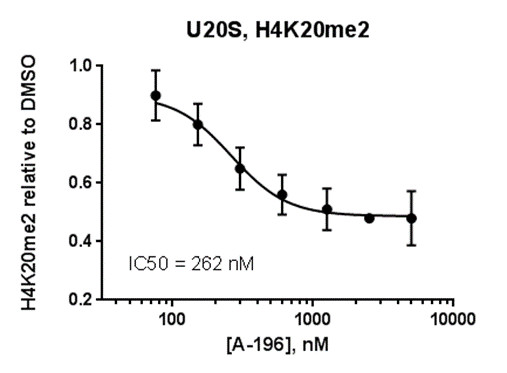
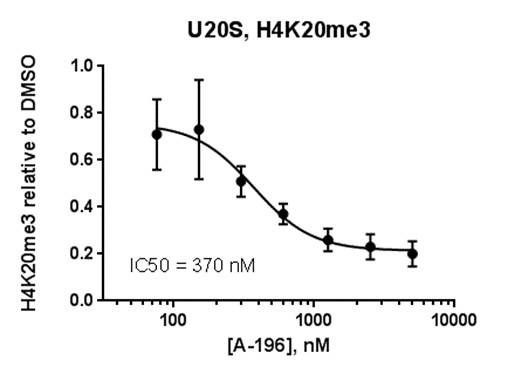
FRAP studies were performed using U20S cells expressing full-length CECR2. Six hours after transfection 2.5 µM SAHA (to increase global histone acetylation) was added and cells were treated with 1 µM or 5 µM of NVS-CECR2-1 1 hour before imaging and half recovery times from the fluorescence signal of the bleached U2OS nuclei were plotted.
NanoBRET
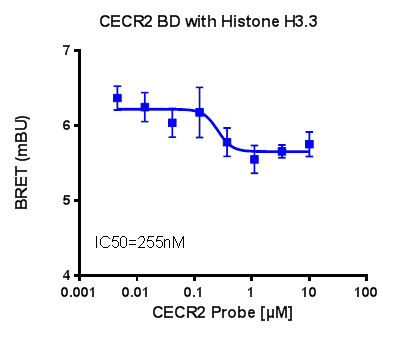
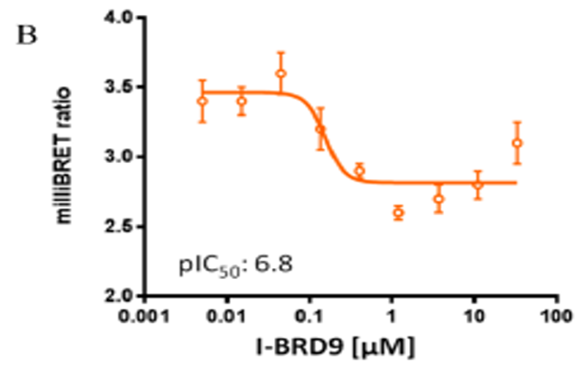
U2OS cells were co-transfected with Histone H3.3-HaloTag and NanoLuc-CECR2. Twenty hours post-transfection cells were collected, washed with PBS, and exchanged into media containing phenol red-free DMEM and 4% FBS in the absence (control sample) or the presence (experimental sample) of 100 nM NanoBRET 618 fluorescent ligand (Promega). Cells were then treated with an increasing dose of NVS-CECR2-1. Five minutes prior to reading, NanoBRET furimazine substrate (Promega) was added to both control and experimental samples and plates were read on a CLARIOstar (BMG) equipped with 450/80 nm bandpass and 610 nm longpass filters with a 0.5 sec reading setting. A corrected BRET ratio was calculated and is defined as the ratio of the emission at 610 nm/450 nm for experimental samples (i.e. those treated with NanoBRET fluorescent ligand) subtracted by and the emission at 610 nm/450 nm for control samples (not treated with NanoBRET fluorescent ligand). BRET ratios are expressed as milliBRET units (mBU), where 1 mBU corresponds to the corrected BRET ratio multiplied by 1000. Relative IC50 values were estimated by non-linear regression analysis of (log) concentration of each inhibitor versus milliBRET ratios (GraphPad Prism).

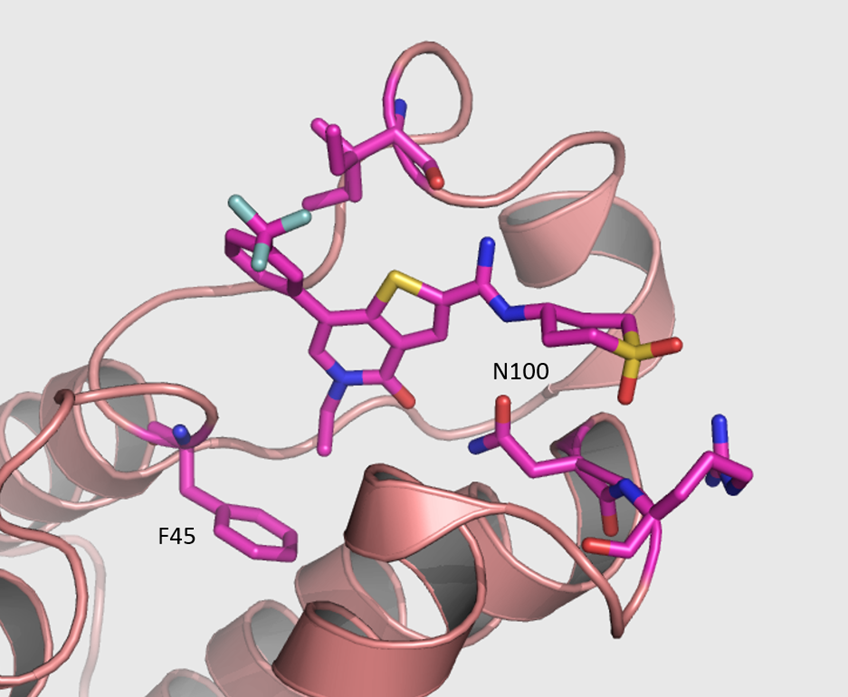
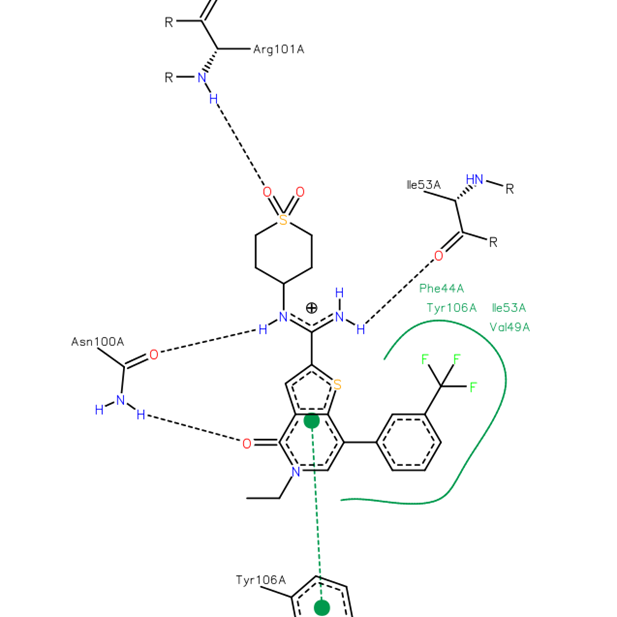
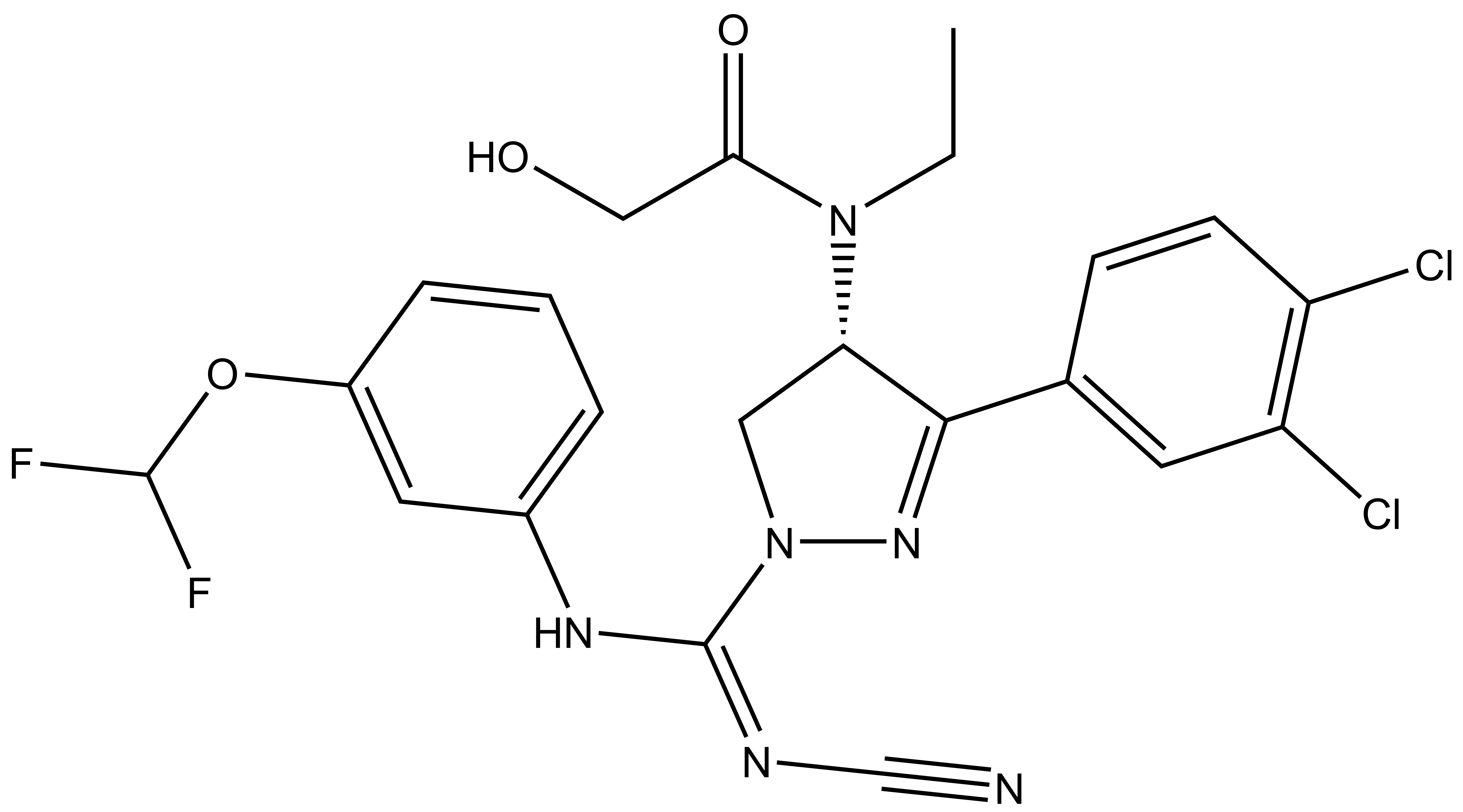
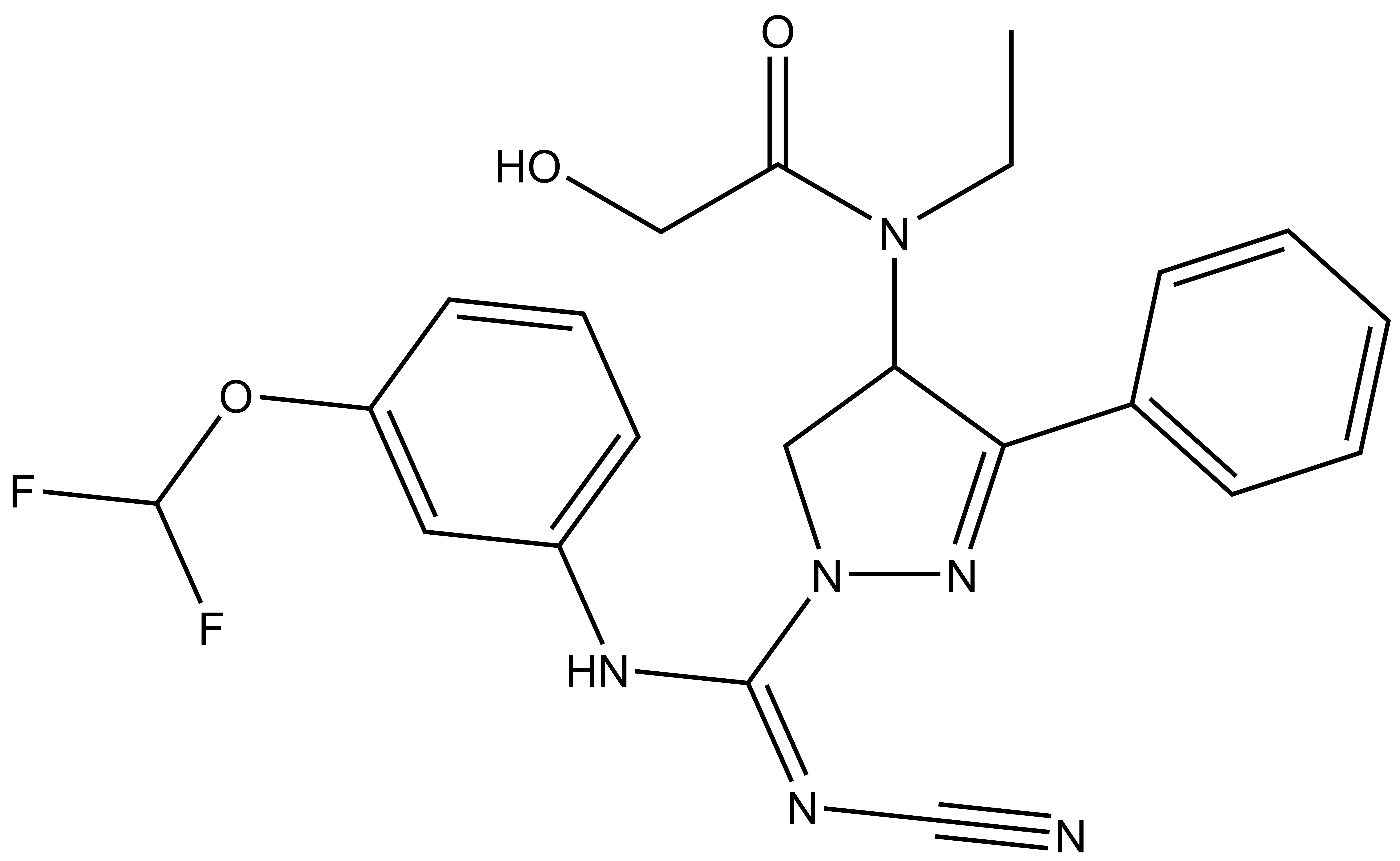
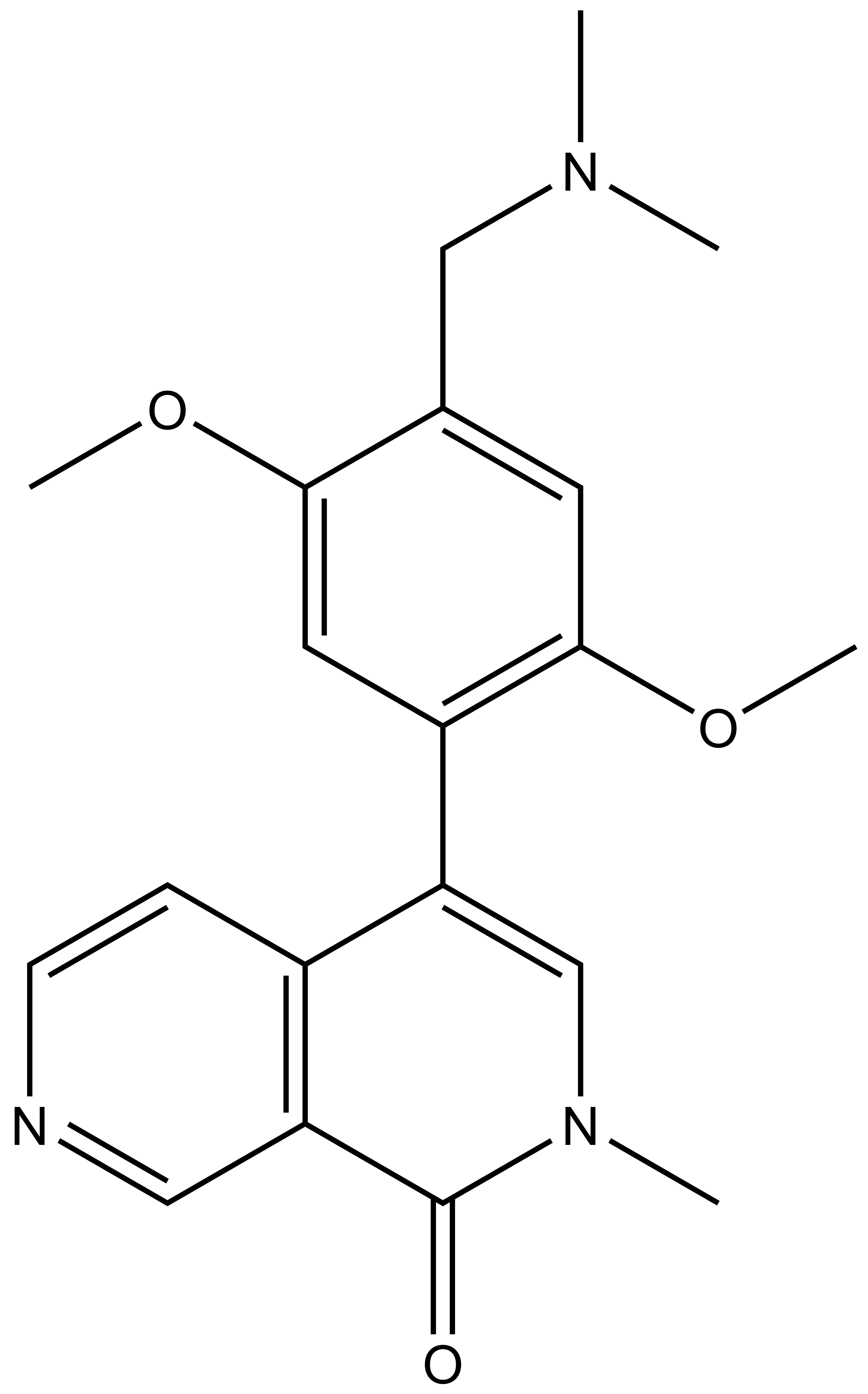
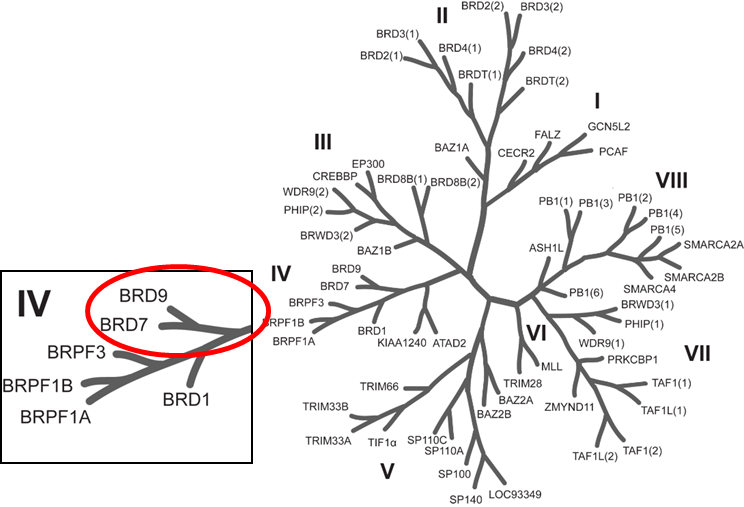
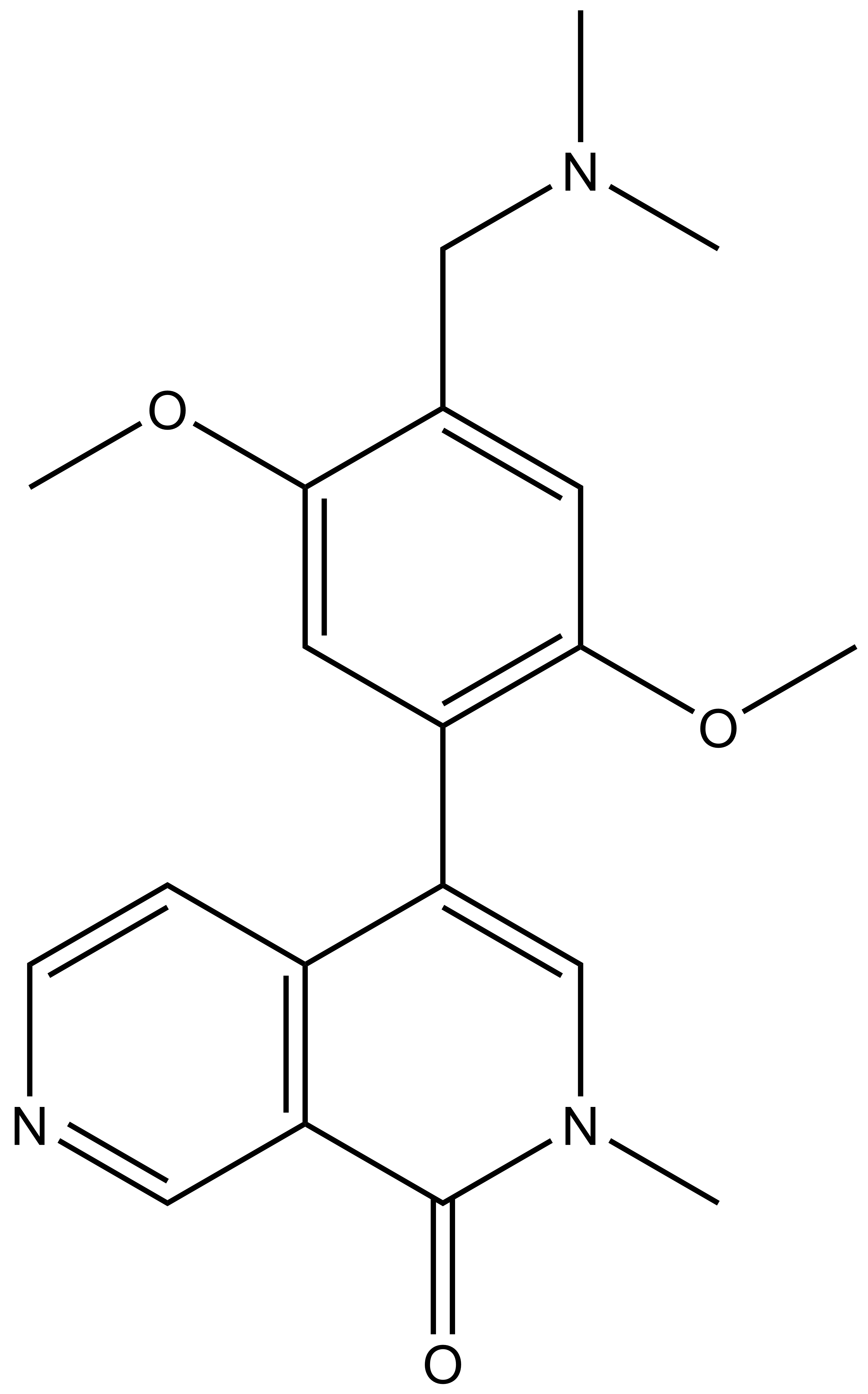
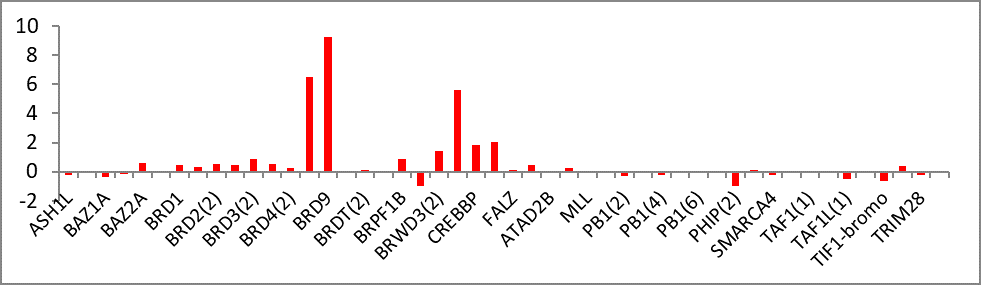
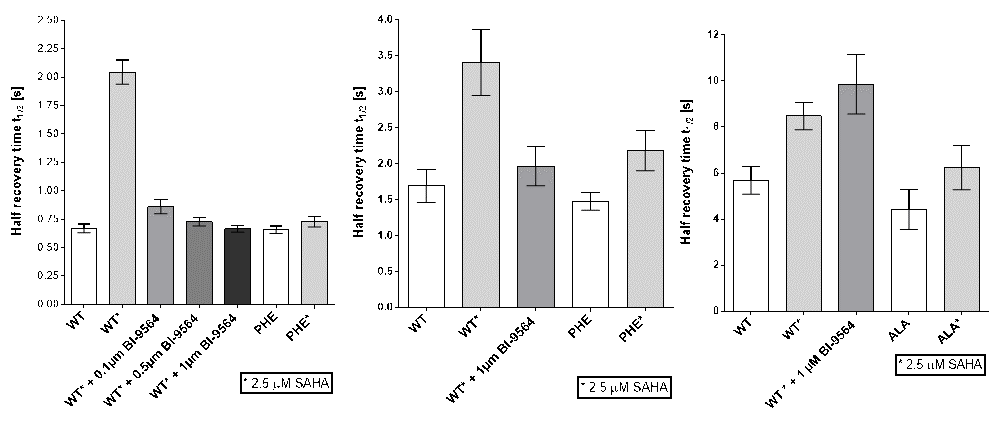
{kind=link}
{kind=link}