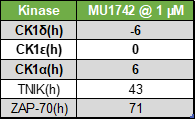
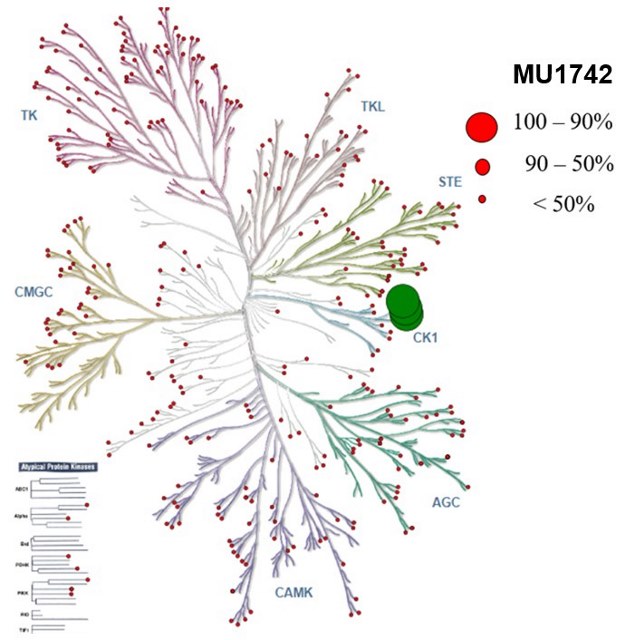
| Probe | | Negative control |
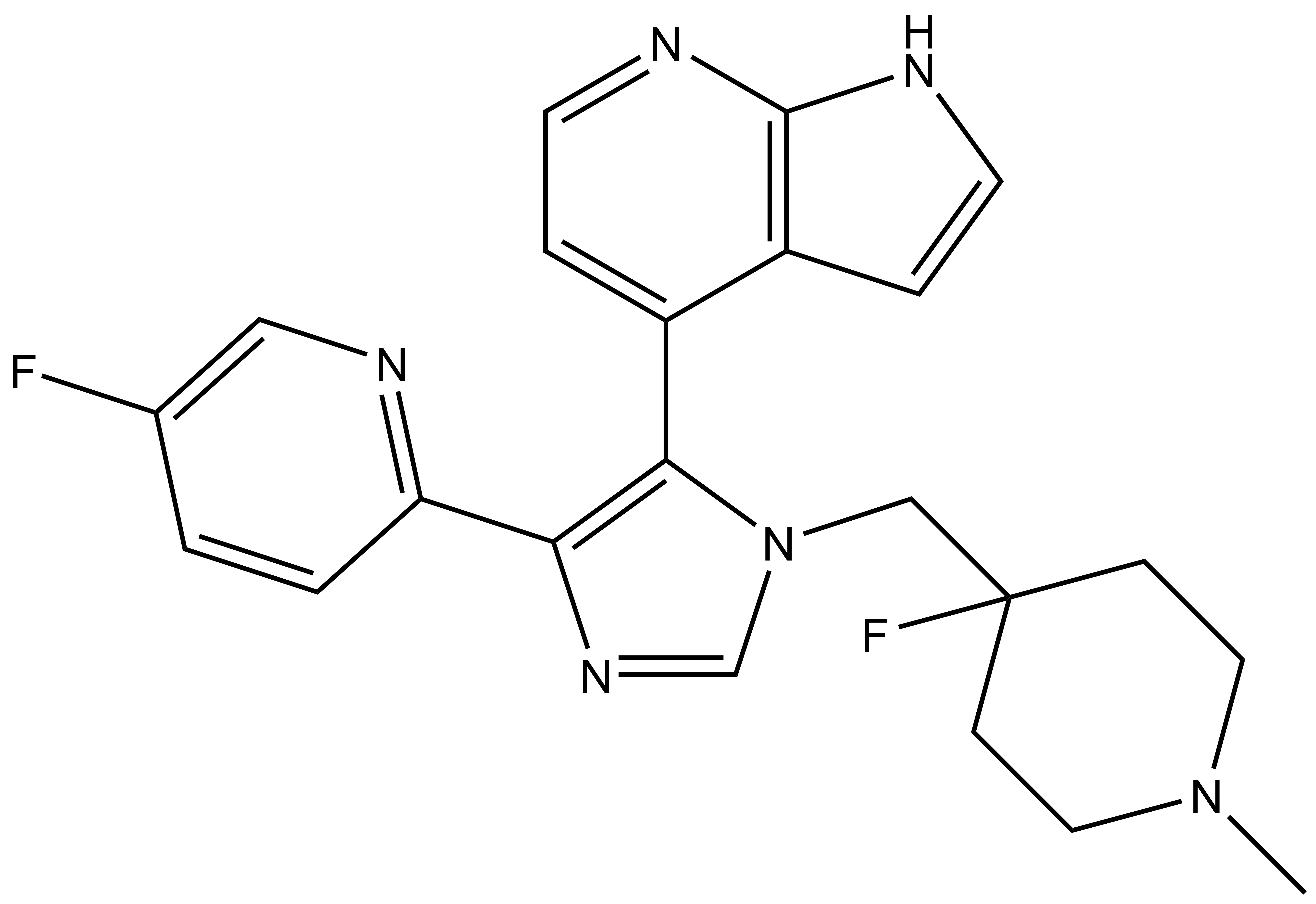 | | 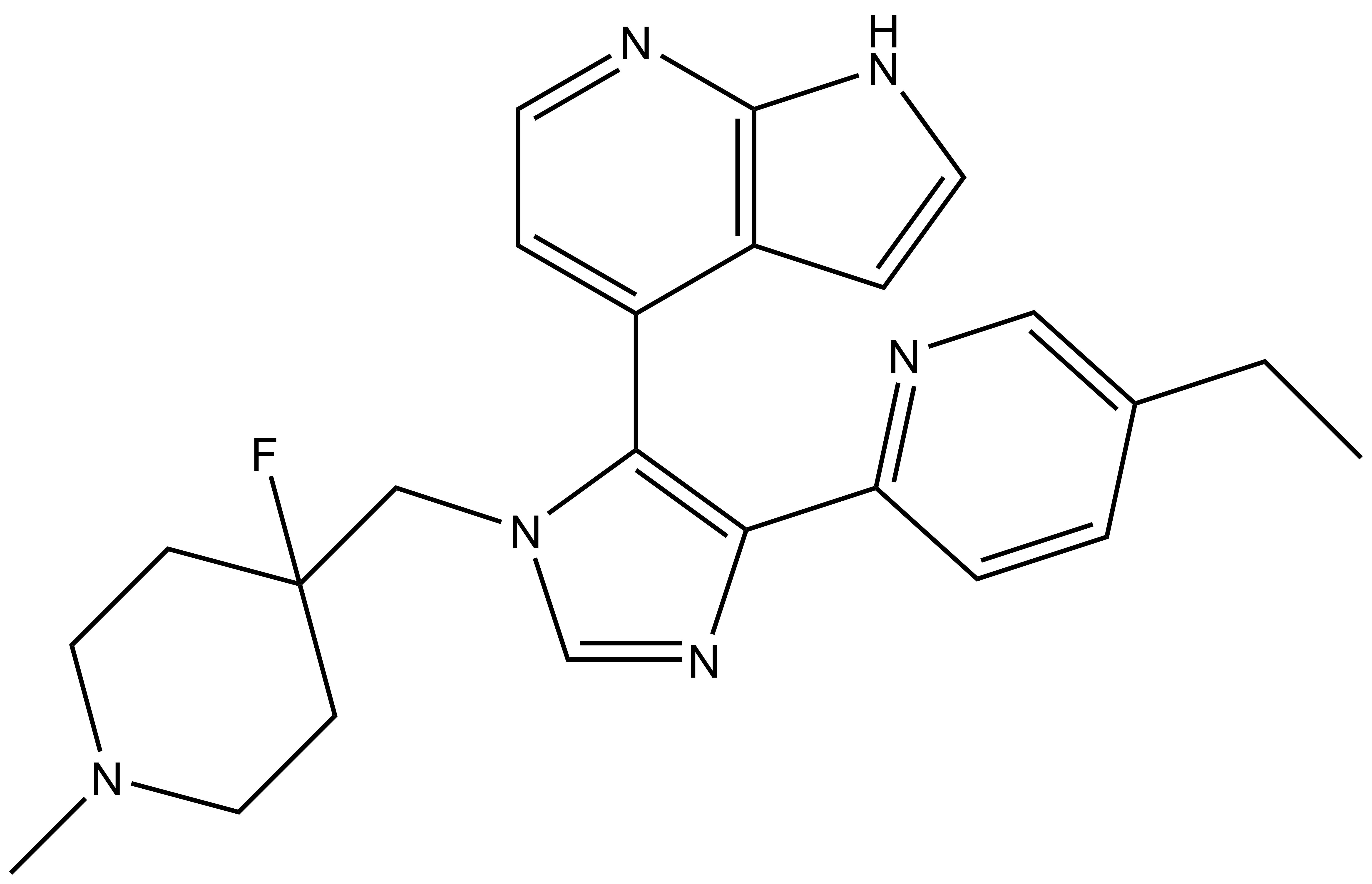 |
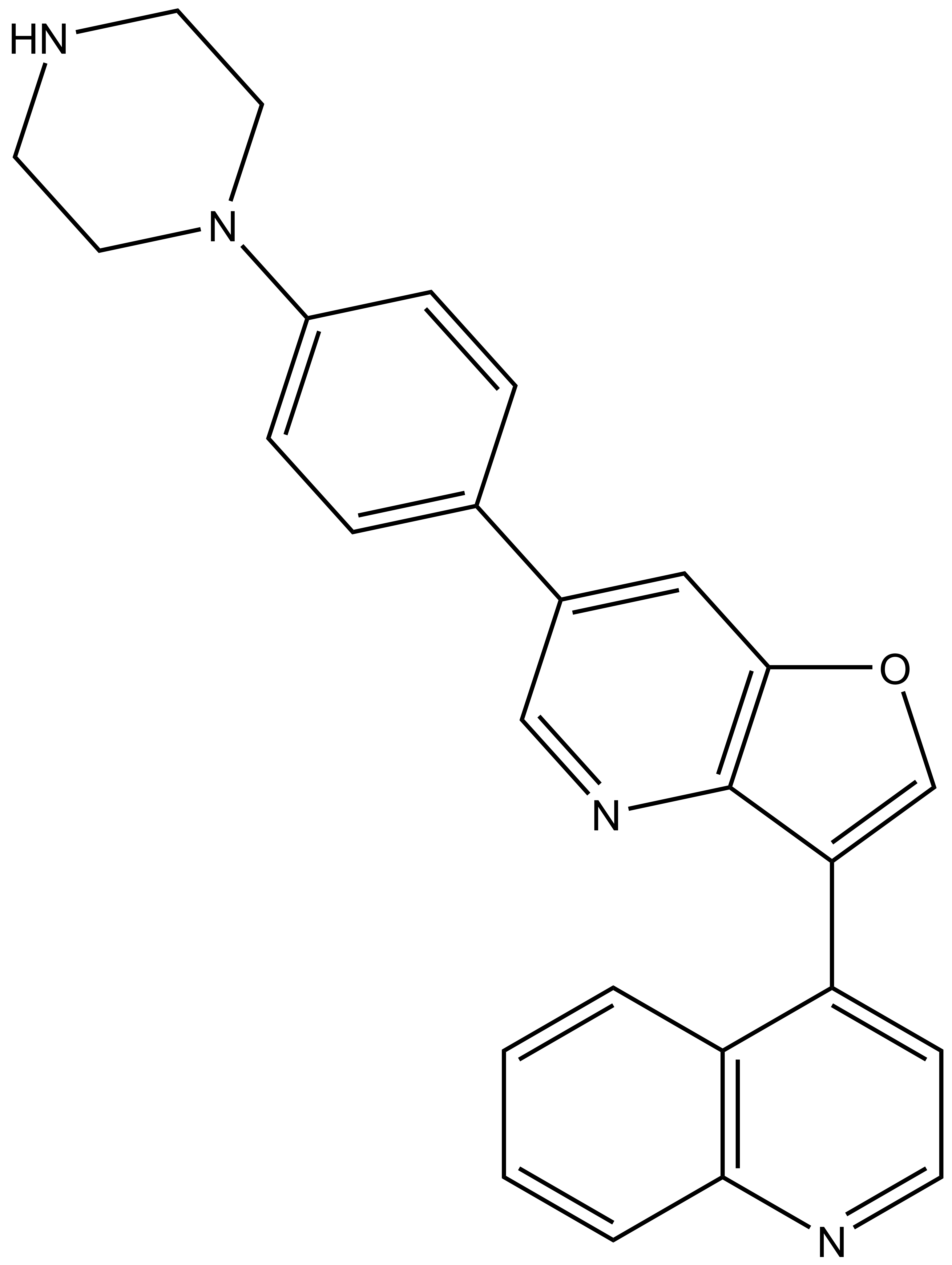
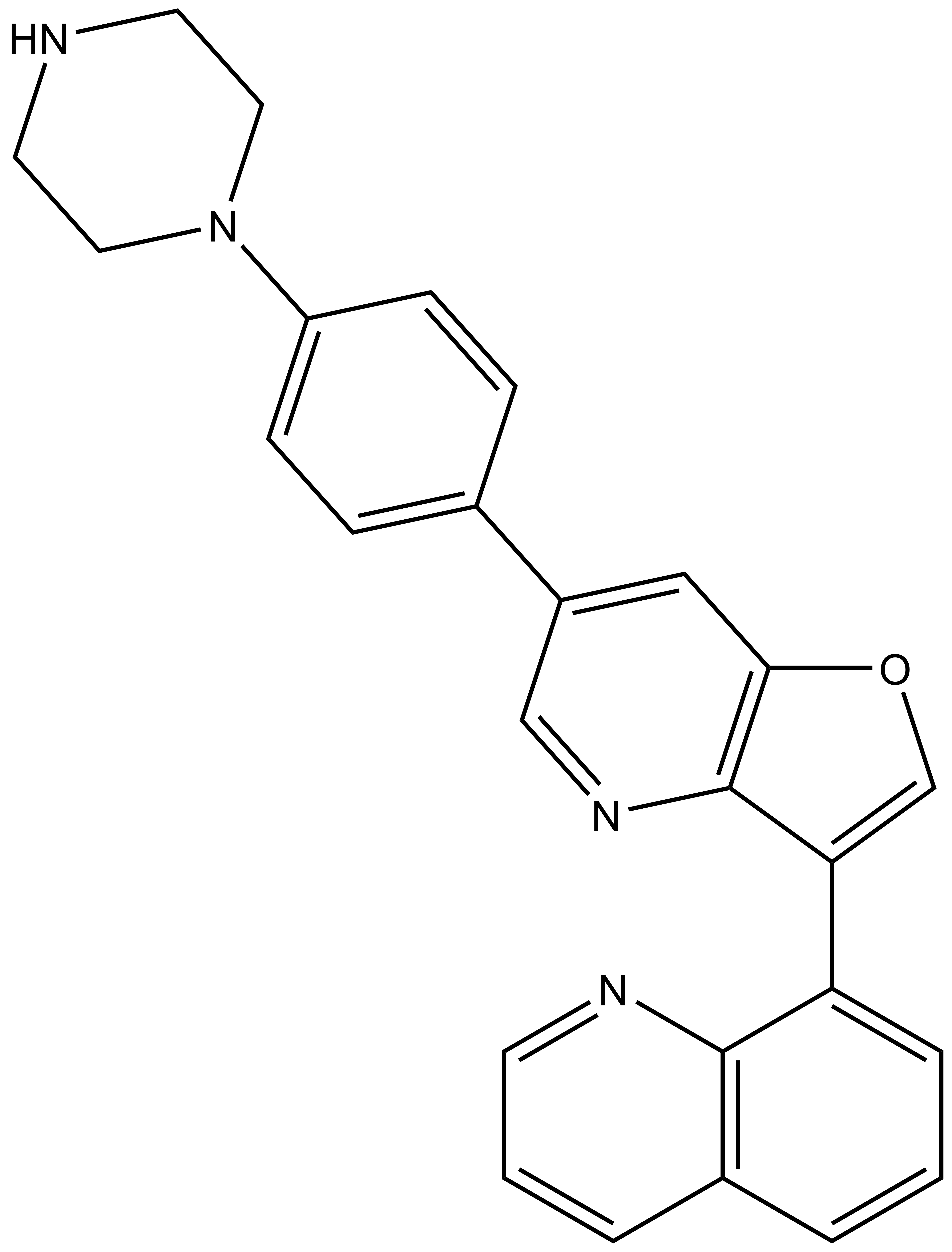
MU1742 | | MU2027 |
Casein kinases 1 (CK1) belong to the family of serine/threonine kinases.1 There are six CK1 isoforms (CK1α, CK1γ1, CK1γ2, CK1γ3, CK1δ, CK1ε) and several splice variants in humans.1,2 All CK1 isoforms share highly conserved kinase domain, with the highest homology displayed by CK1δ and CK1ε.3 They are generally cofactor-independent, and depend on N-terminal acidic and/or phosphorylated amino acids for substrate recognition.4 The cellular activity of CK1 can be regulated by sub-cellular localization, post-translational modifications or interaction with other proteins. The major post-translational modifications comprise activating and inhibitory phosphorylation which can be performed site-selectively by other kinases or via autophosphorylation.1,3 For instance, inhibitory autophosphorylation at C-terminus has been described for all human CK1 isoforms.1,3 It was found that CK1δ forms a dimer, which could possibly have negative regulatory effect on the CK1δ kinase activity in vivo.3 Substrate recognition motifs for CK1 kinases are widely distributed in cellular proteins and more than 140 substrates for CK1 isoforms have been reported, indicating their pleiotropic character.3 CK1 regulates Wnt, Hh and Hippo pathways, which are important for growth, development and homeostasis.3,5,6 Thus, CK1s are involved in the regulation of various cellular states, processes and functions such as chromosome segregation, gene expression, cellular morphology, immune response and inflammation, membrane trafficking, cytokinesis, autophagy, cell stemness and differentiation, cell survival, proliferation and apoptosis.1–3,7 Although CK1 isoforms are similar in their structure and function (especially CK1δ/ε), they have also distinct and specific functions.
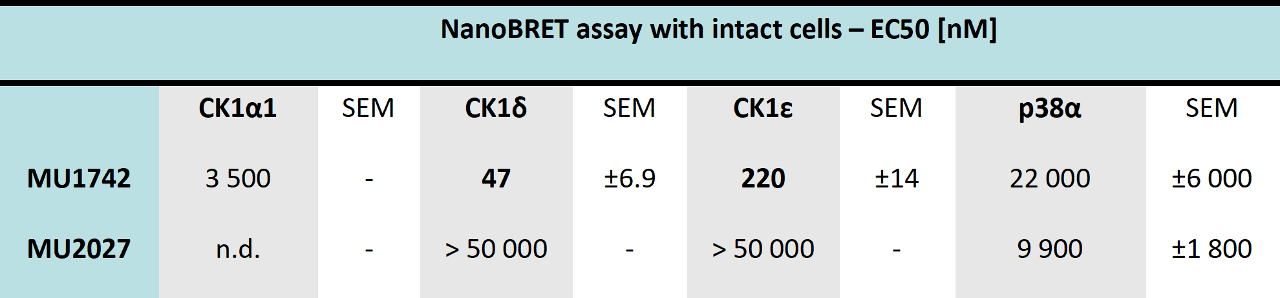
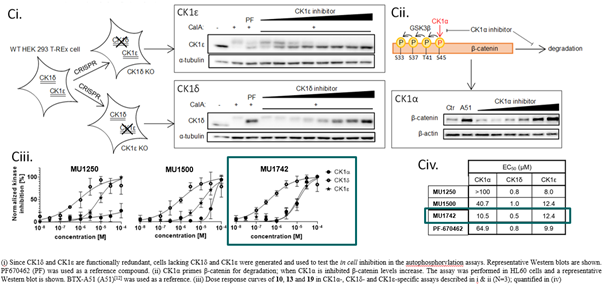
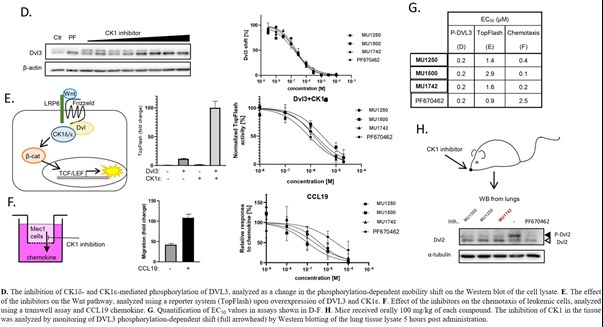
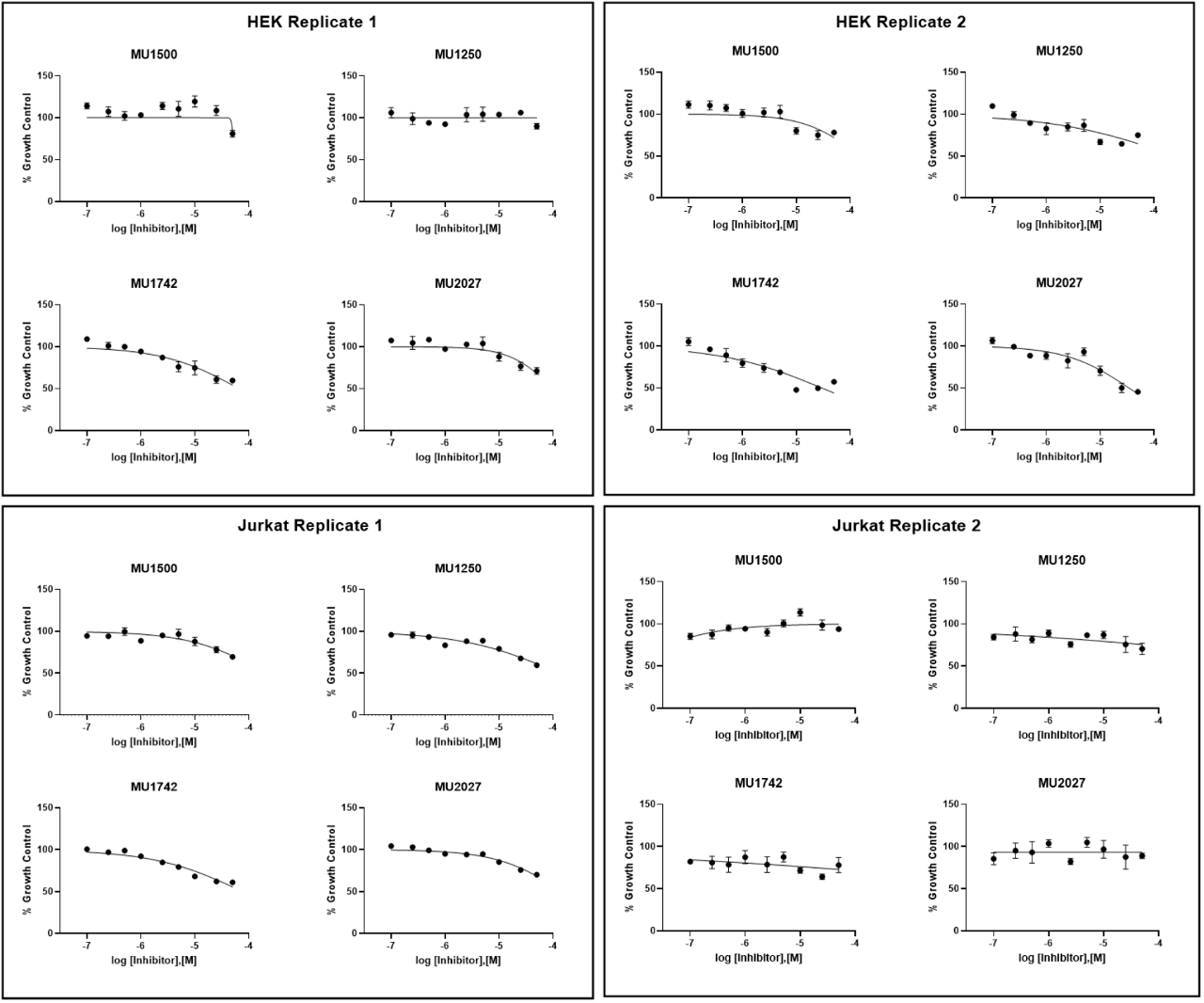
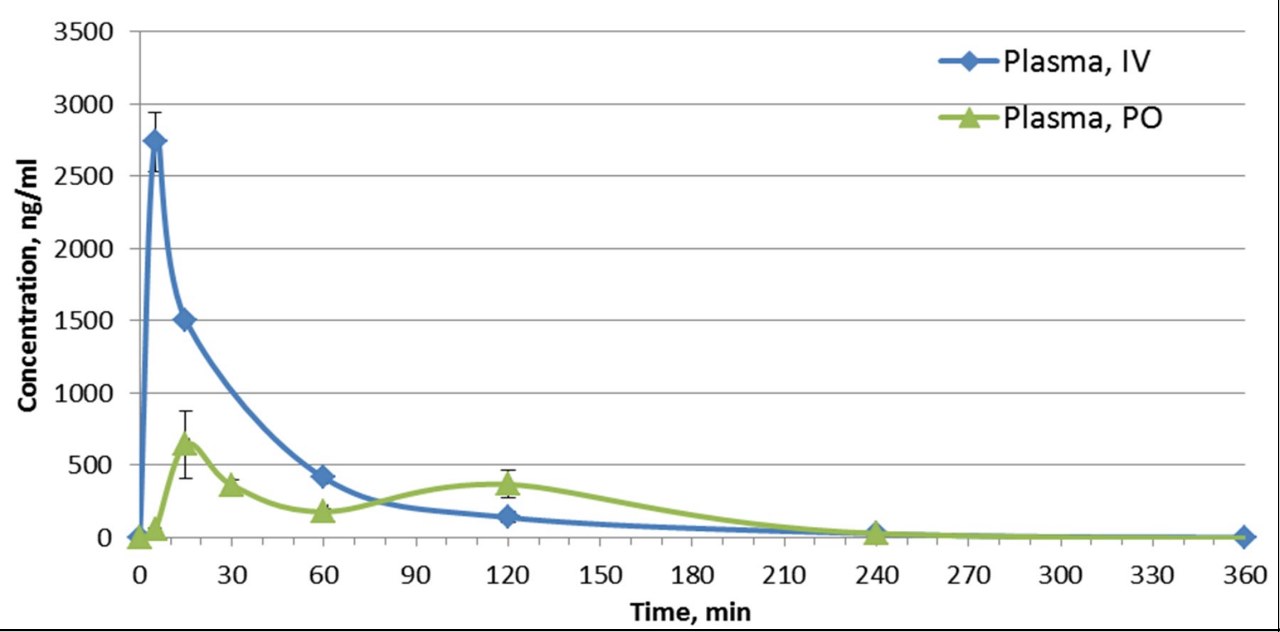
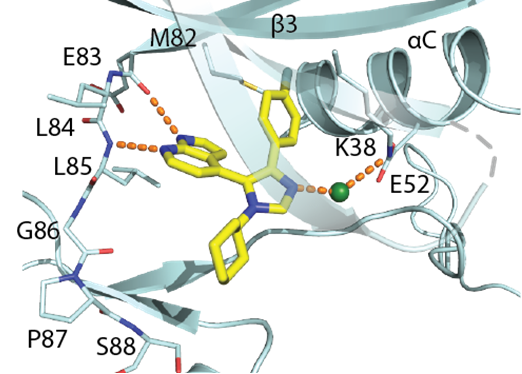
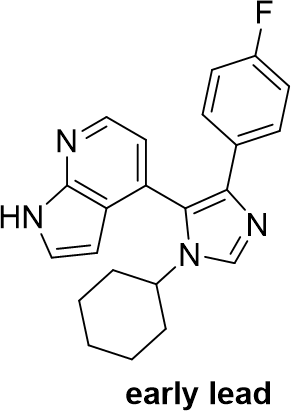
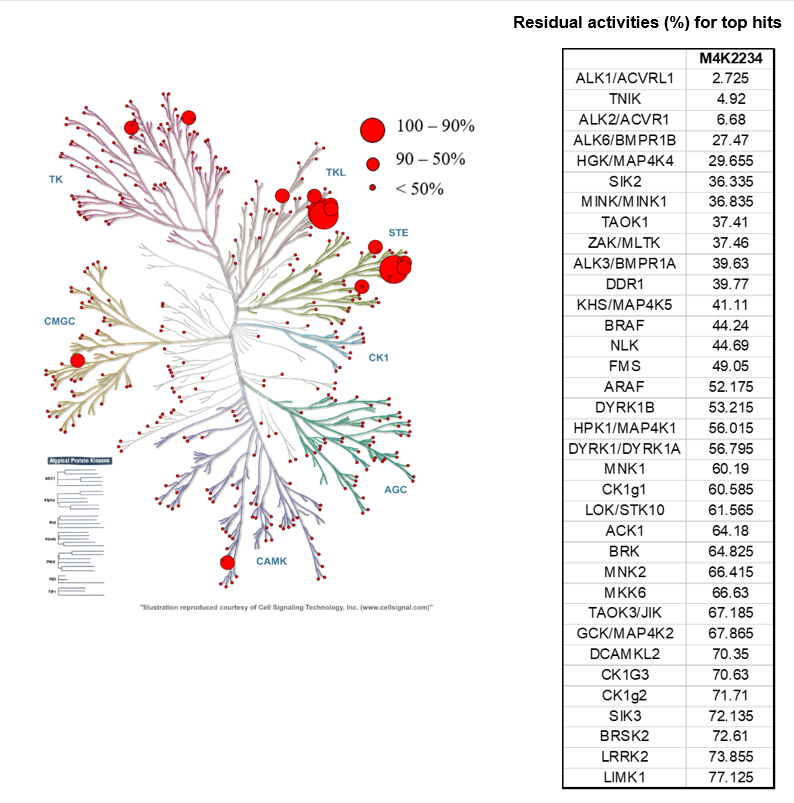
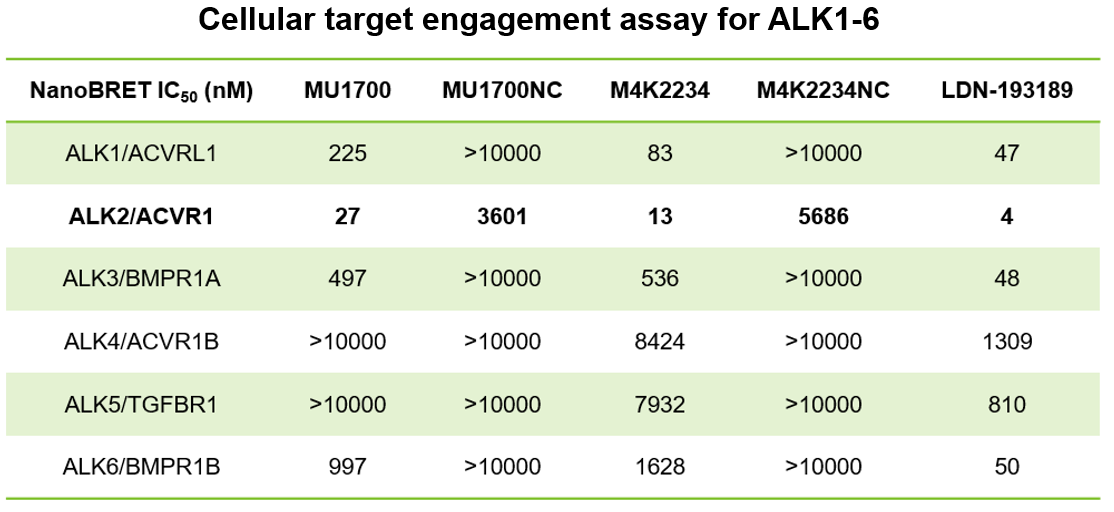
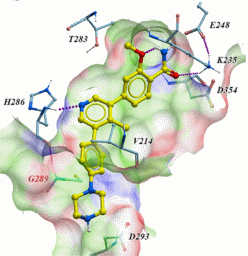
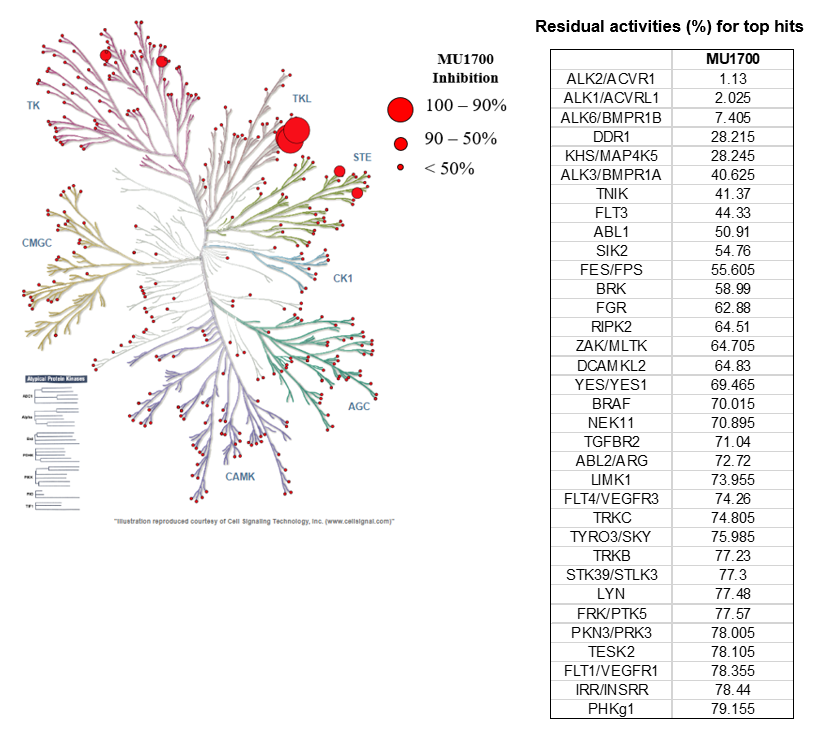
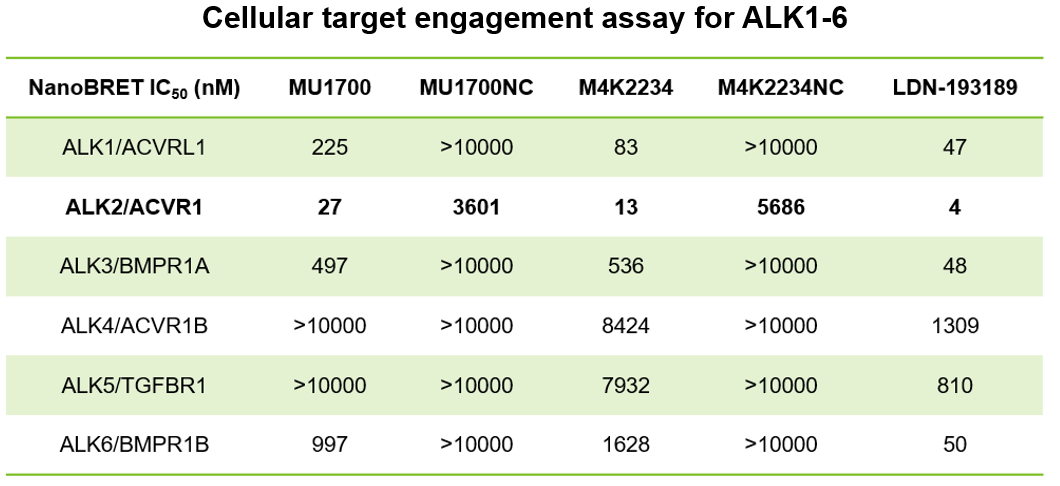
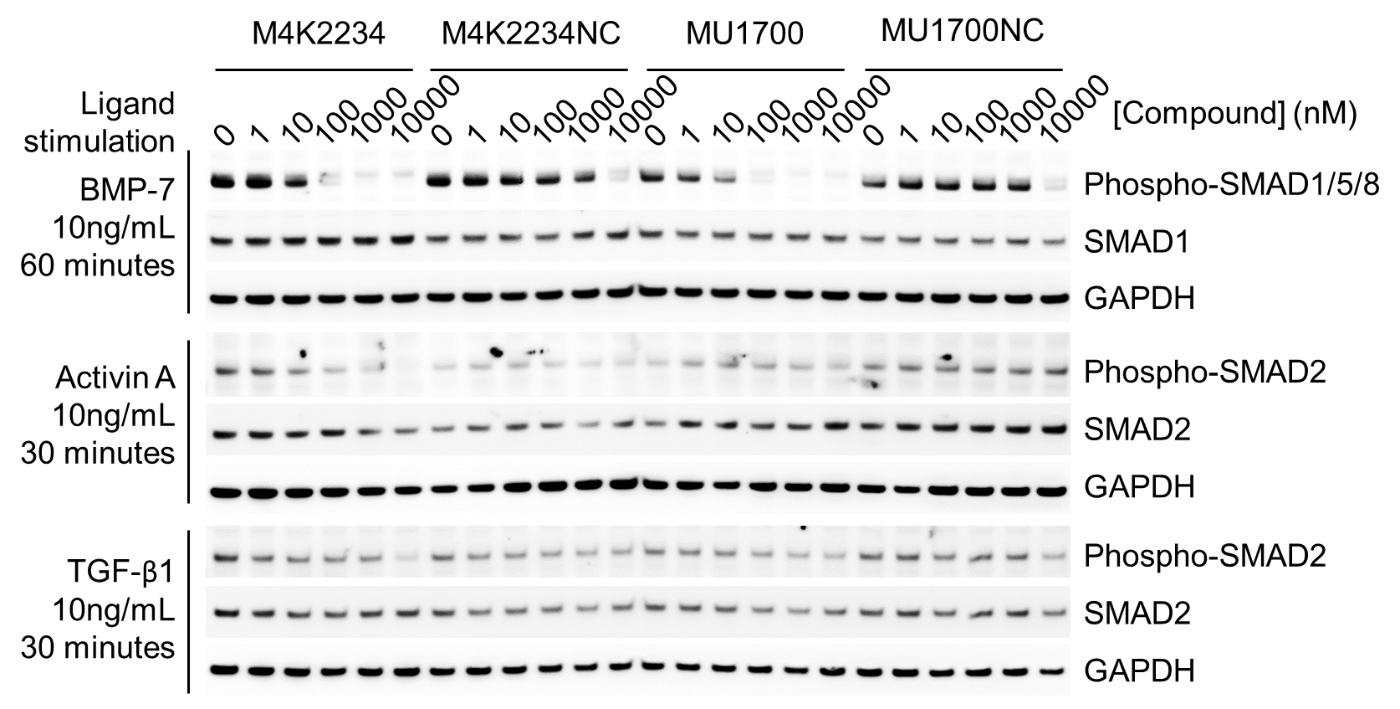
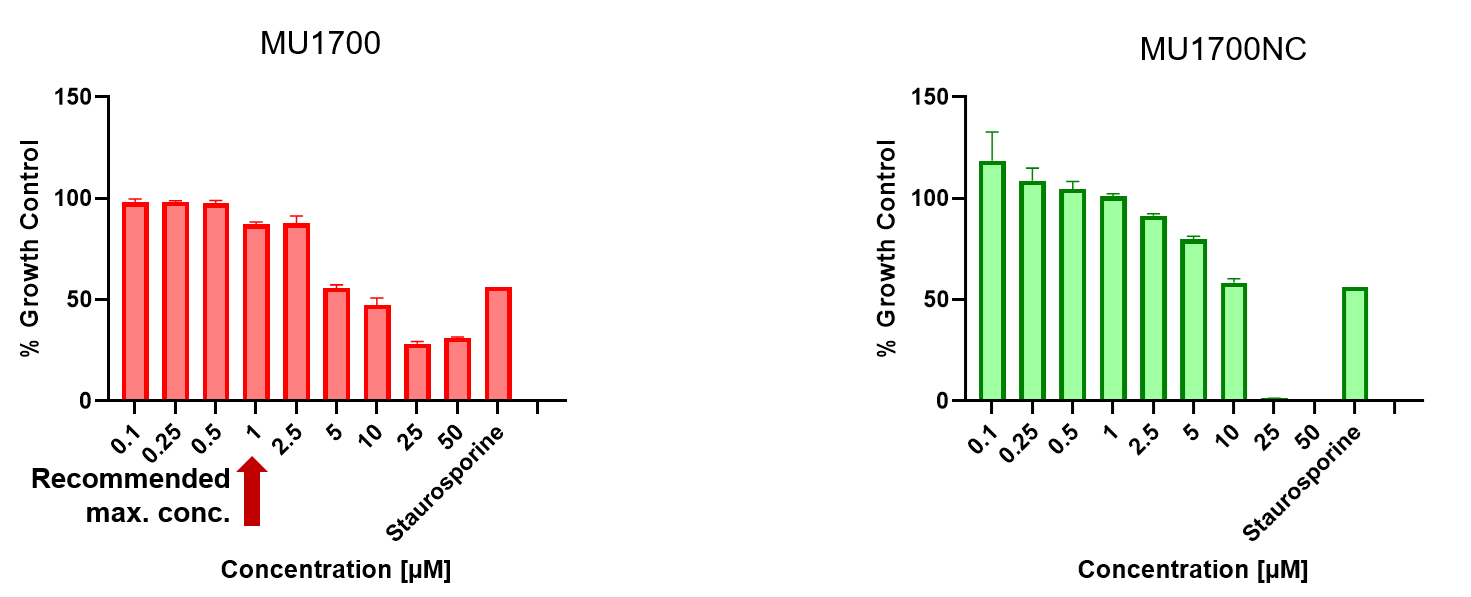
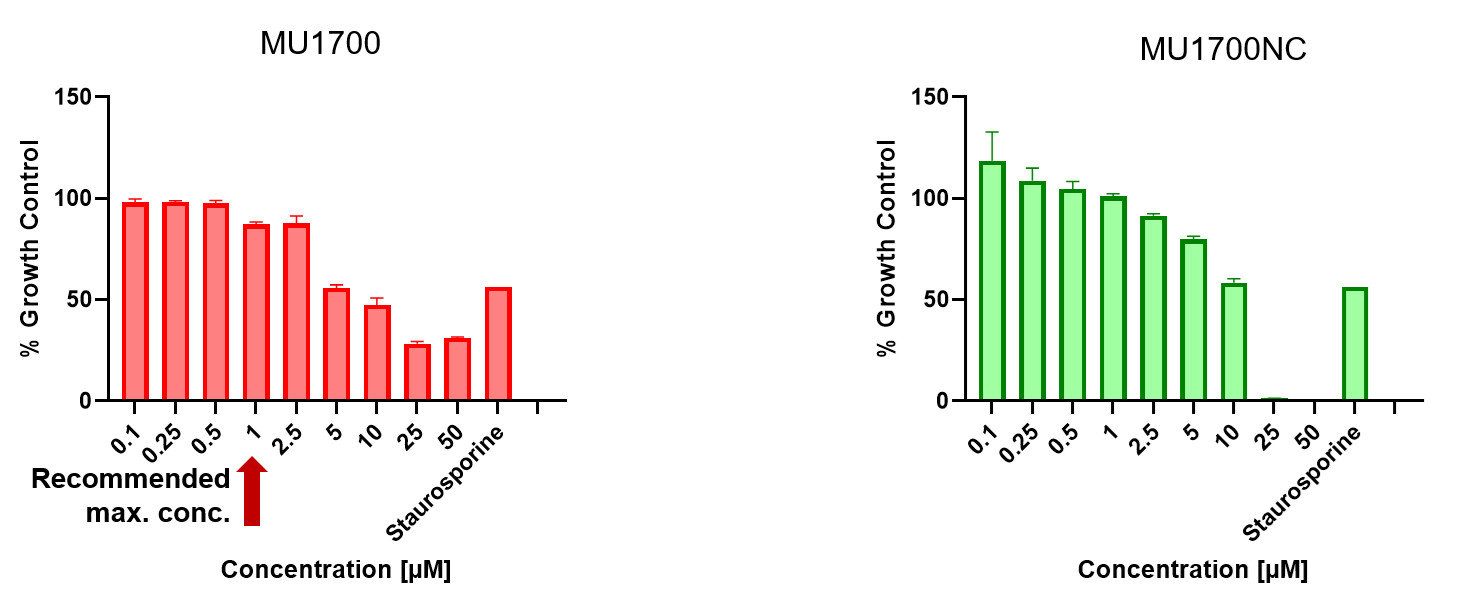
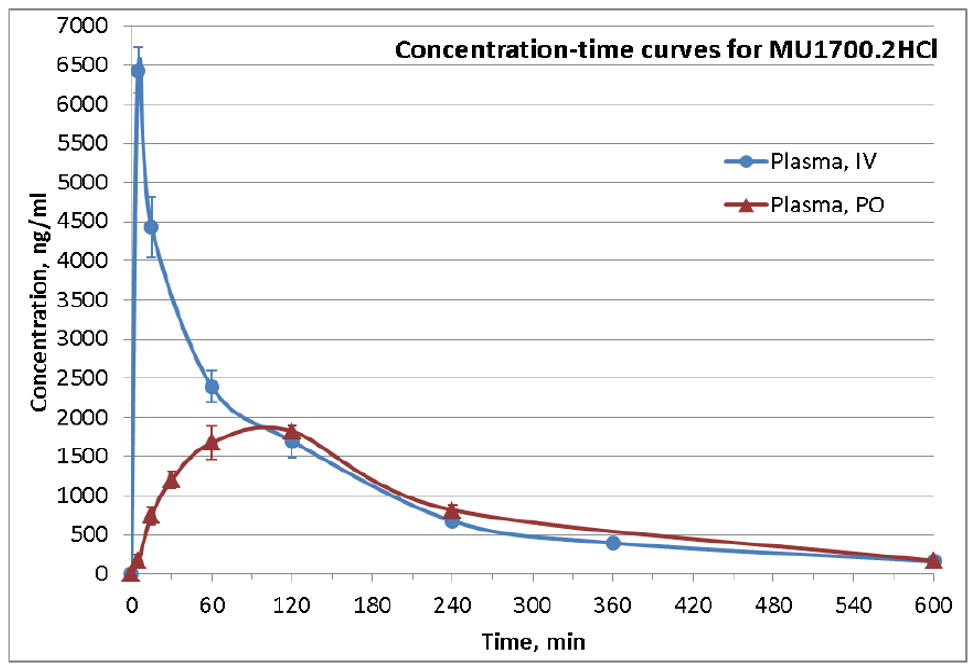
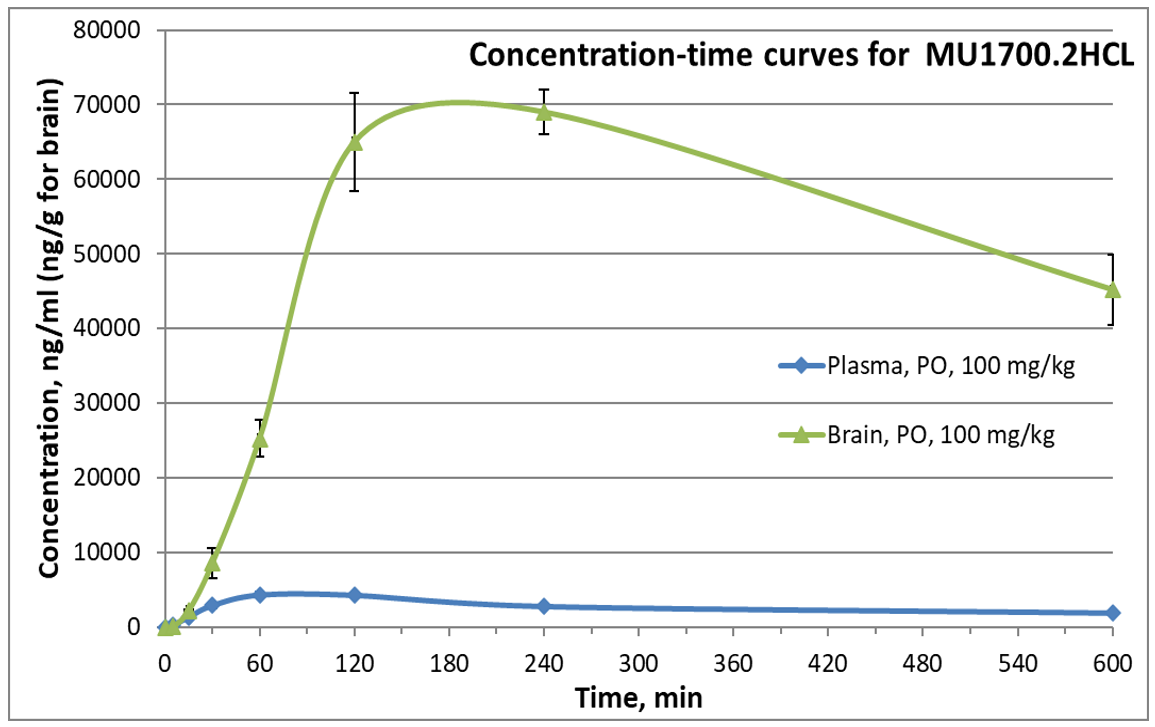
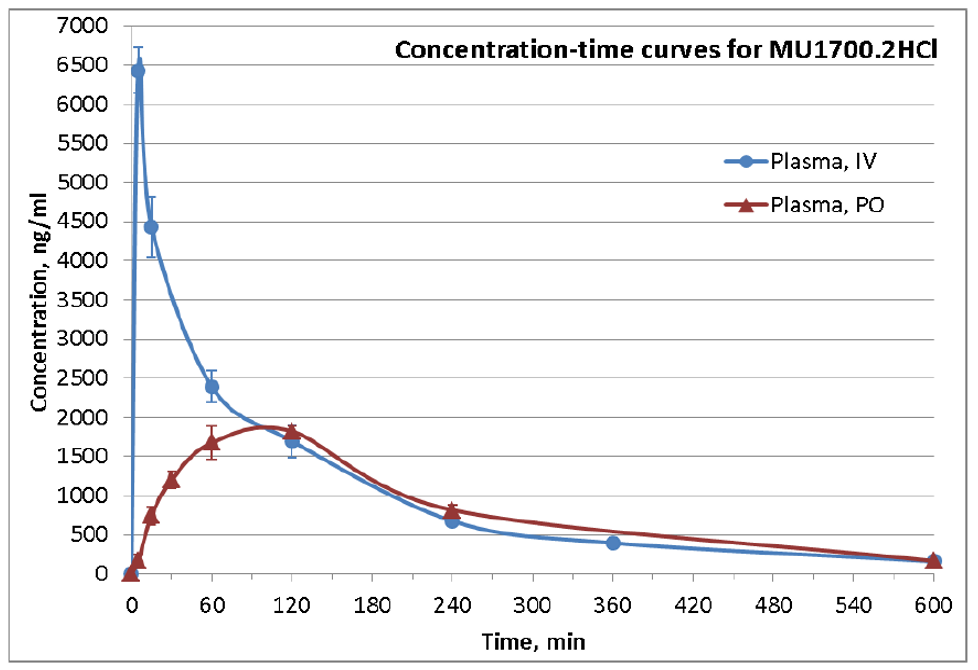
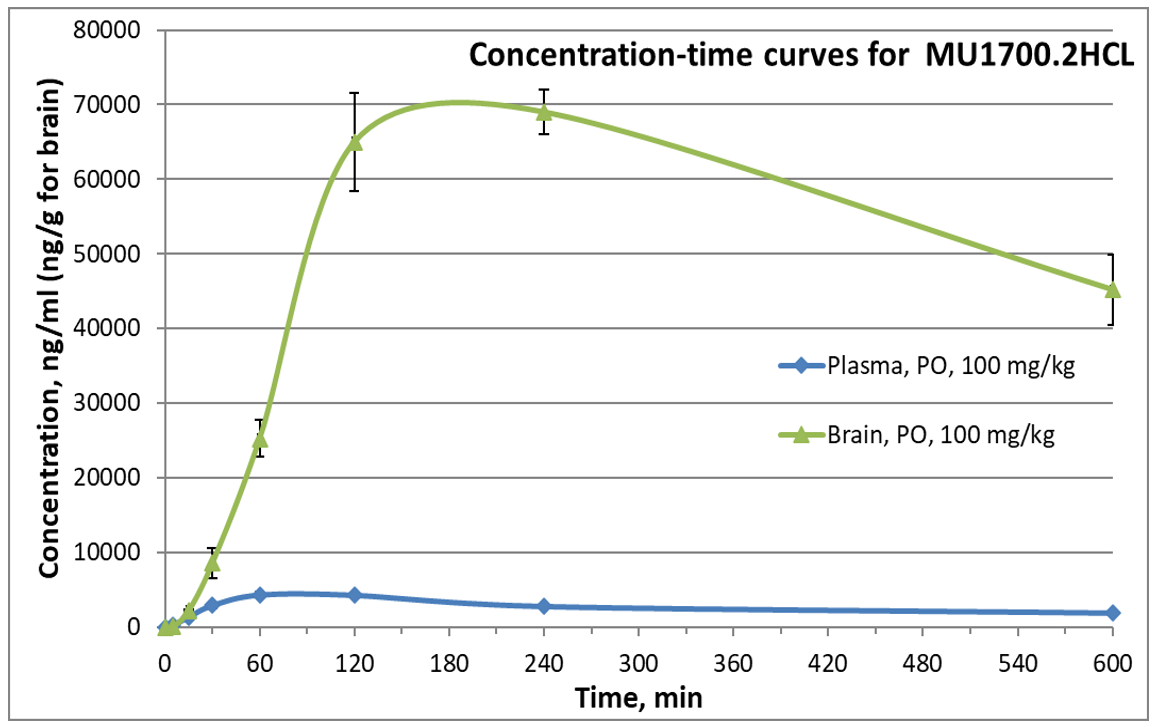
SGC has developed in collaboration with Prof. Kamil Paruch (Masaryk University, Brno, Czech Republic) and Prof. Vitezslav Bryja (Masaryk University, Brno, Czech Republic) quality chemical probe MU1742 for CK1δ and CK1ε protein kinases, including the corresponding negative control compound MU2027. The chemical probe MU1742 exhibits excellent kinome-wide selectivity, high potency against CK1δ/ε in vitro and in cellulo. Moreover, MU1742 has suitable PK profile which allows utilization in vivo.