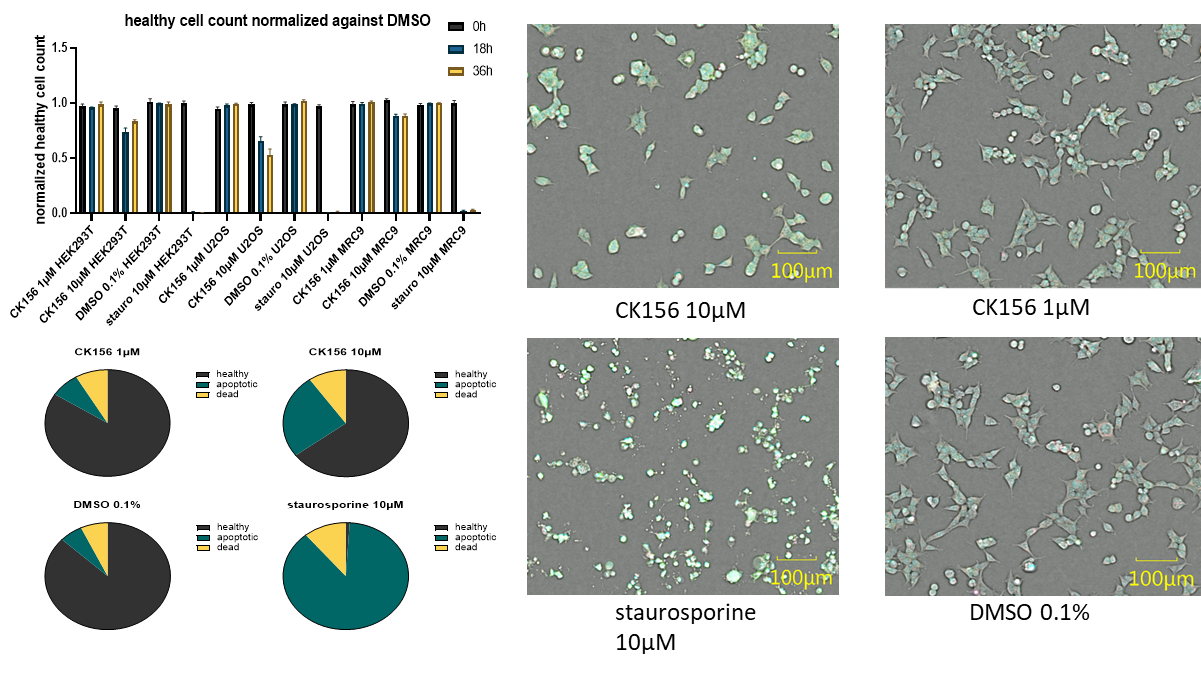
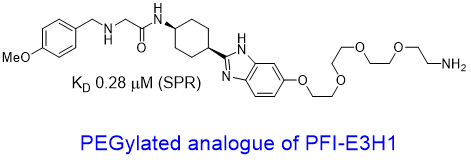
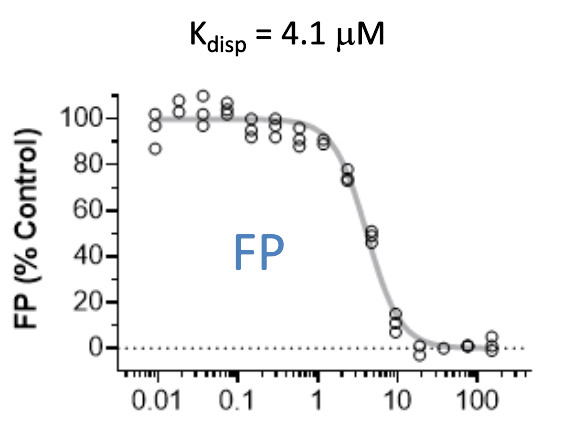
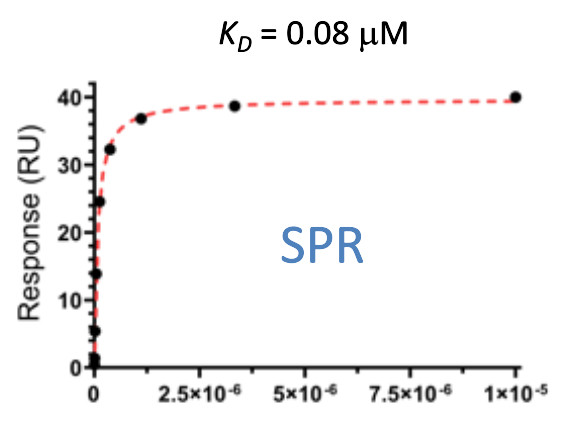
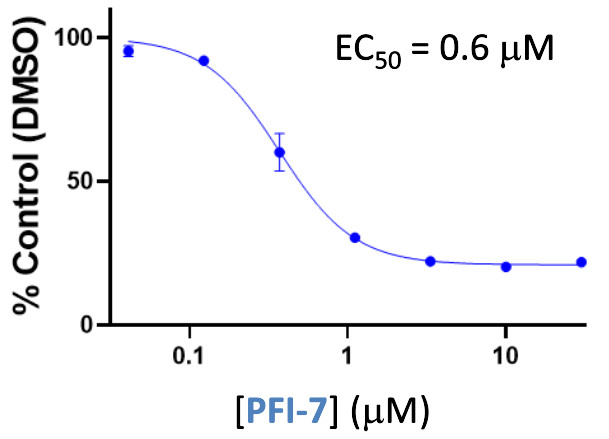
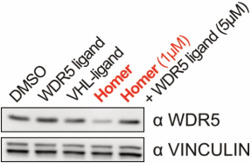
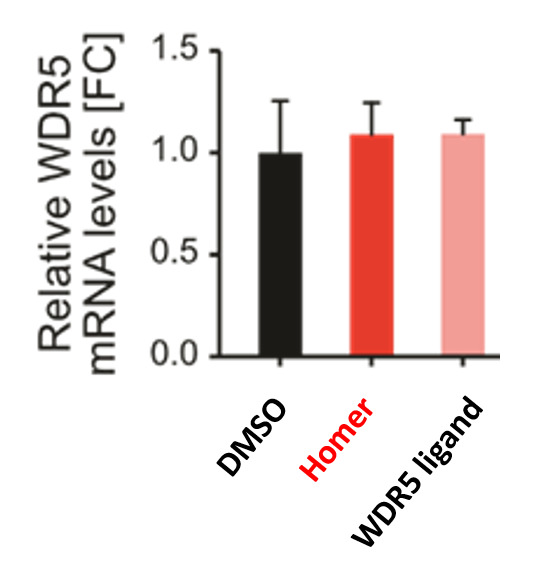
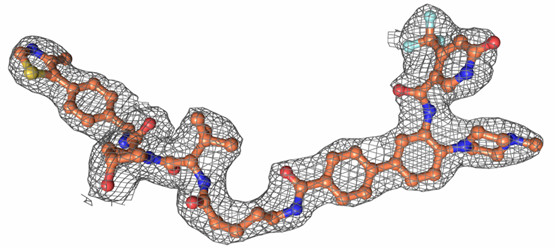
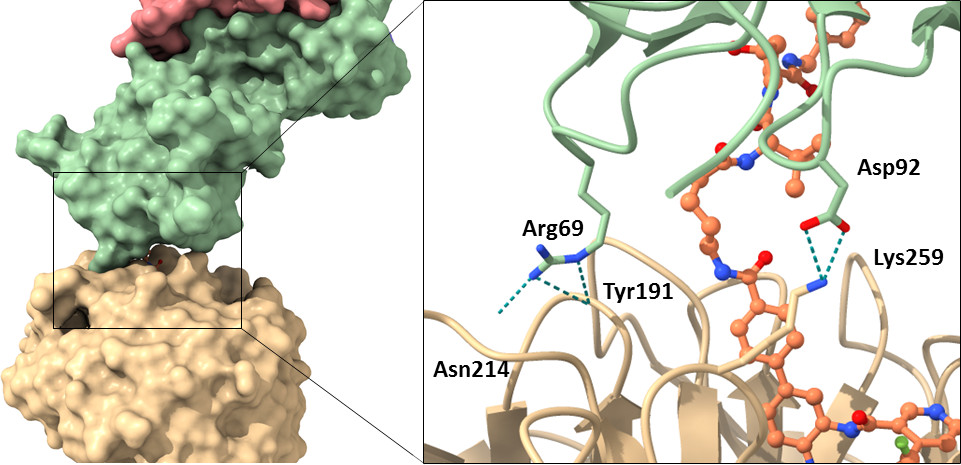
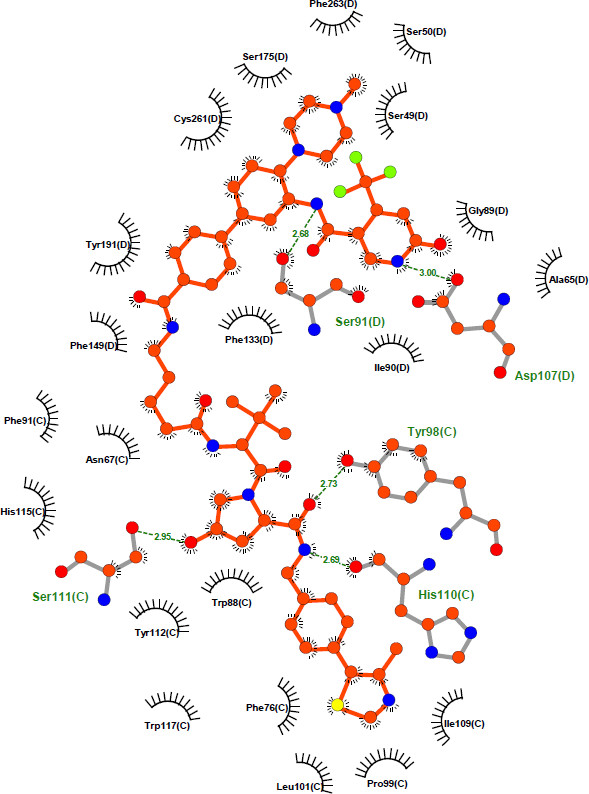
| Probe | | Negative control | | Negative control |
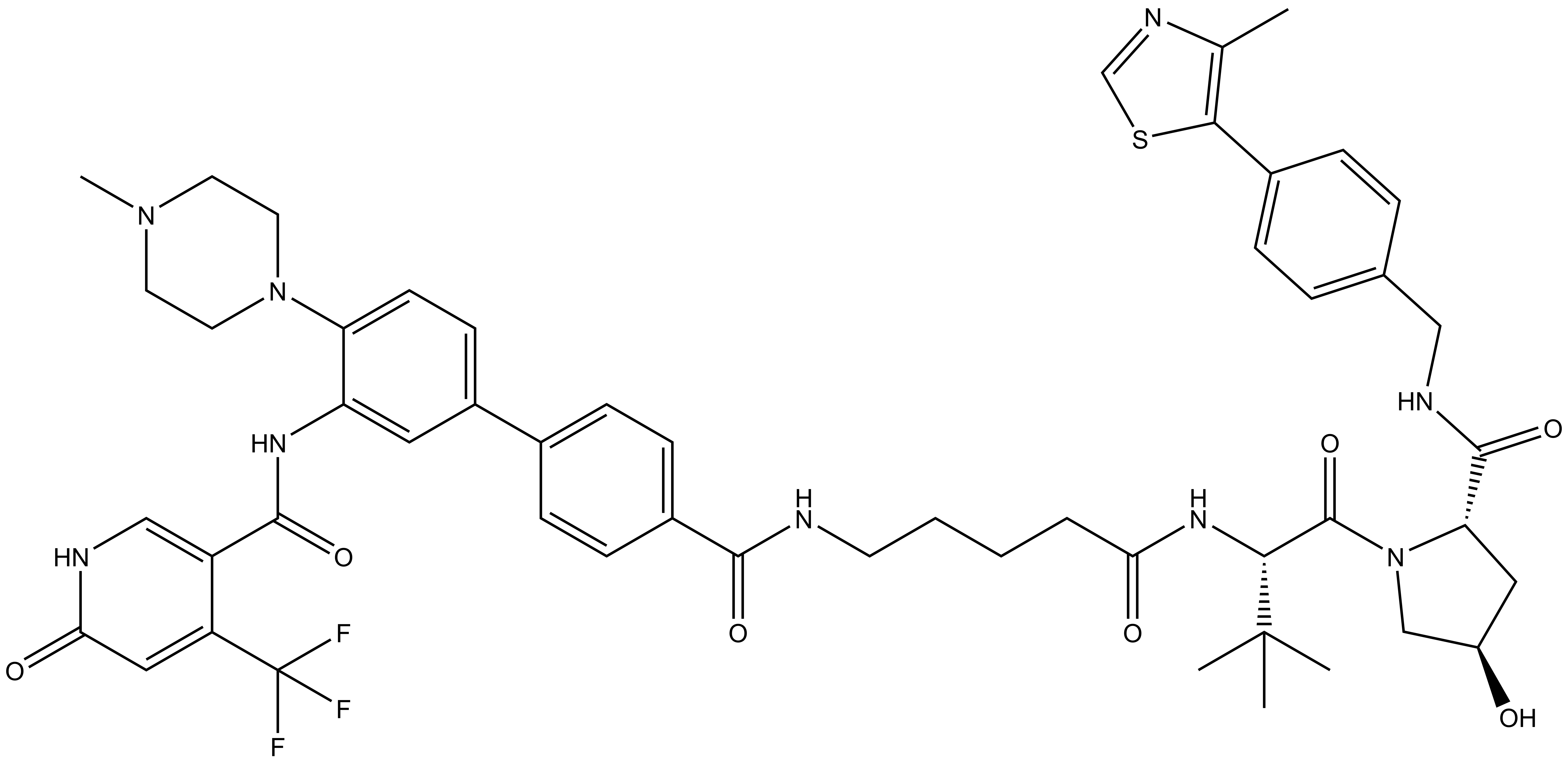 | | 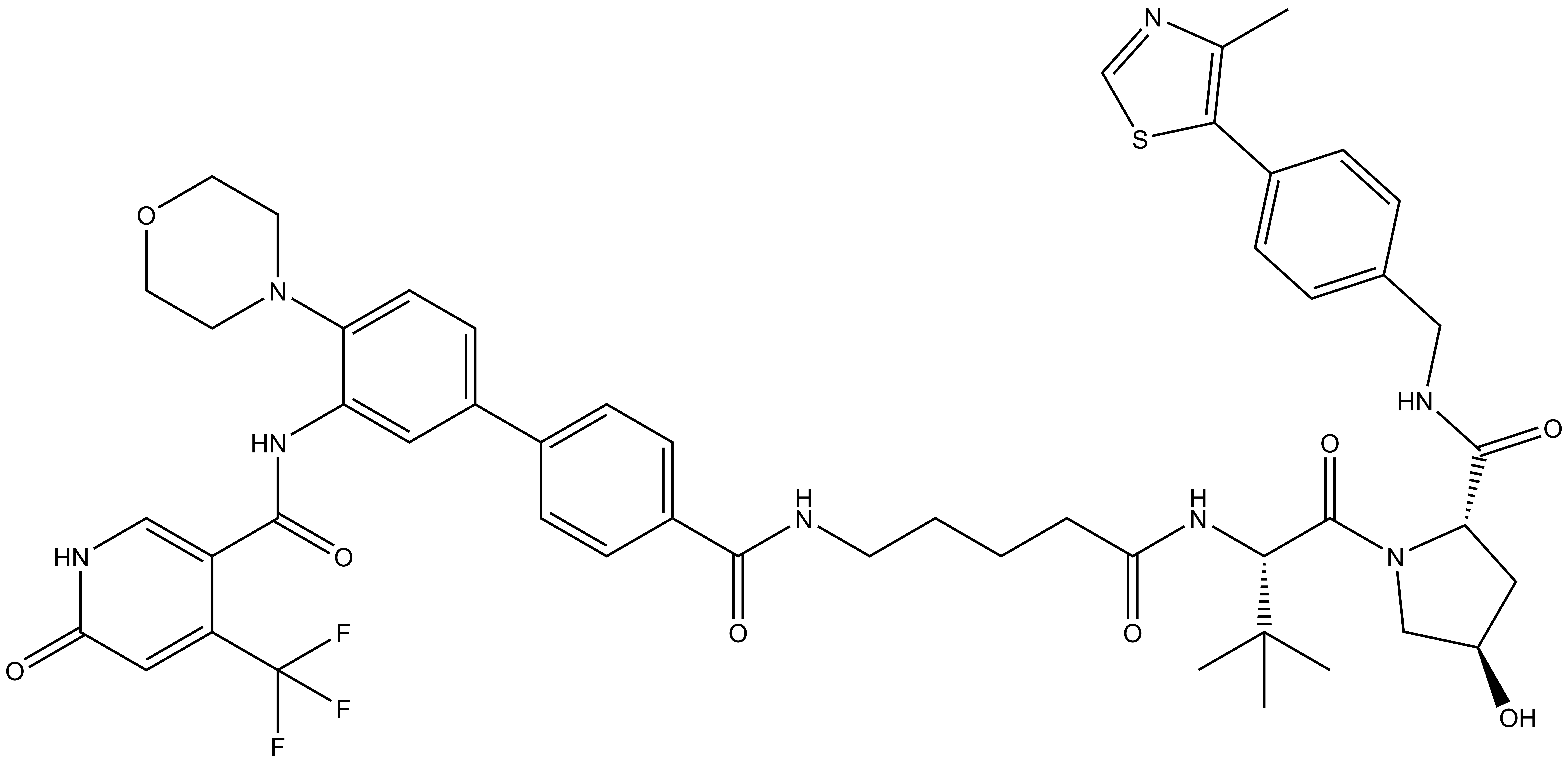 | |  |
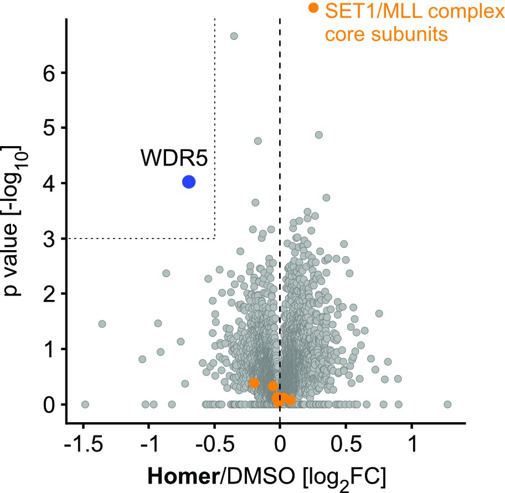
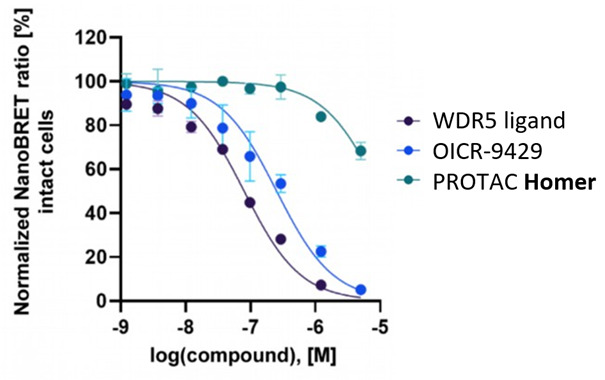
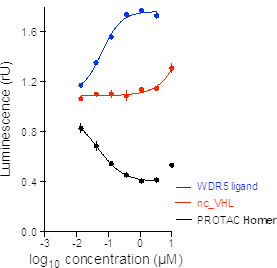
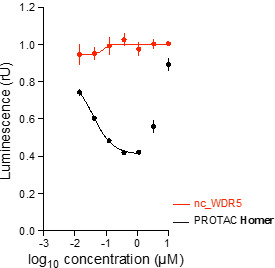
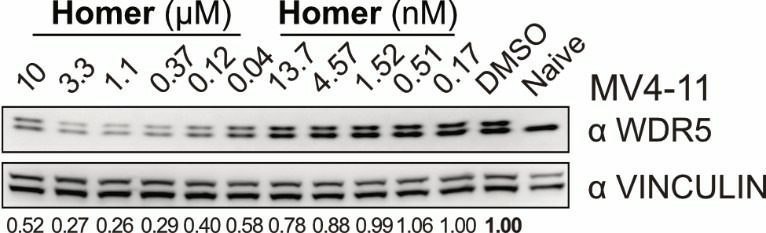
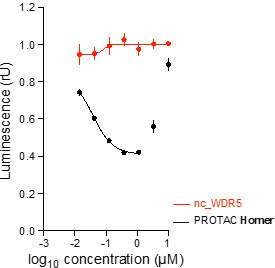
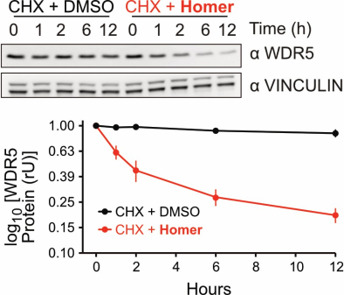
Homer | | nc_WDR5 | | nc_VHL |
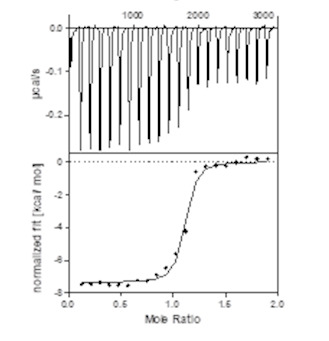
| Homer |
| Physical and Chemical Properties |
| Molecular weight | 1012.16 |
| Molecular formula | C52H60F3N9O7S |
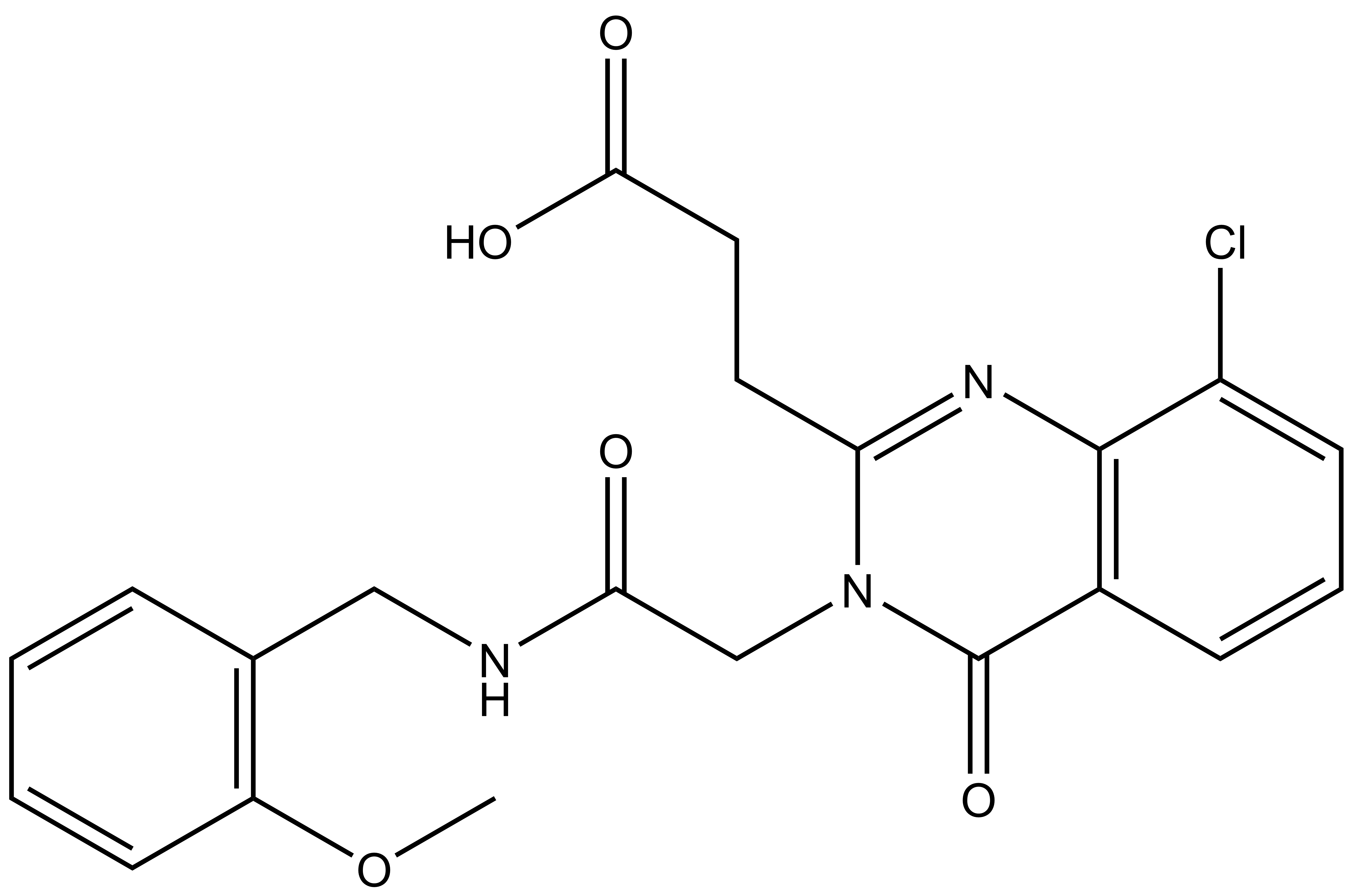
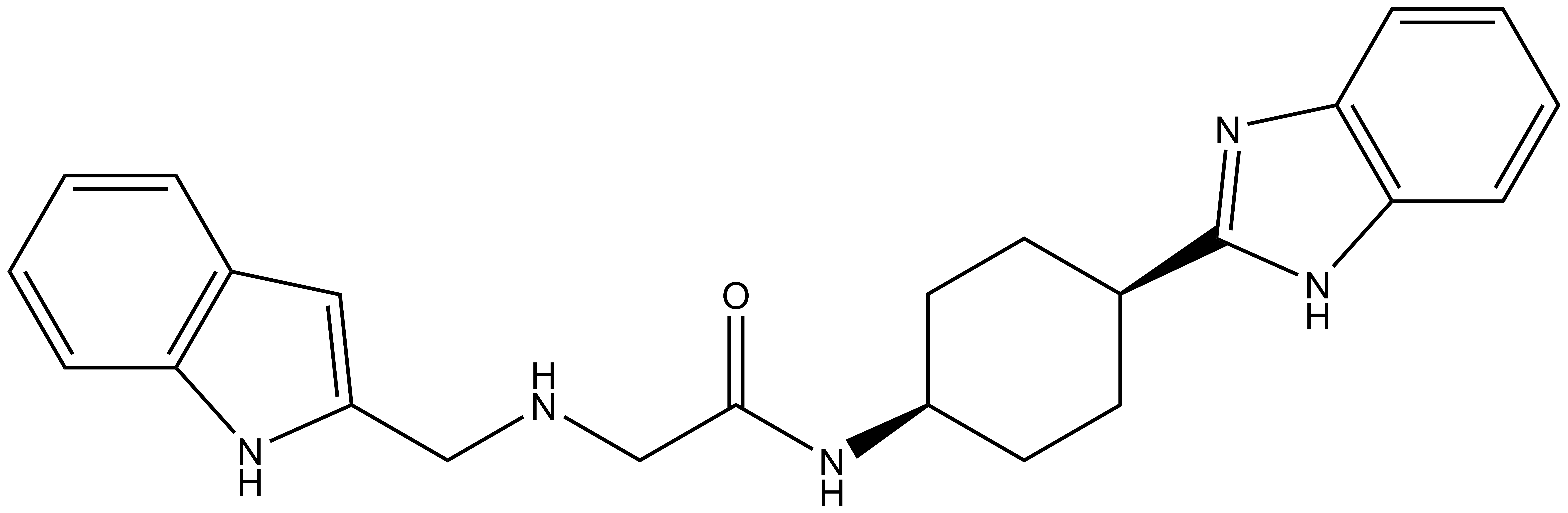
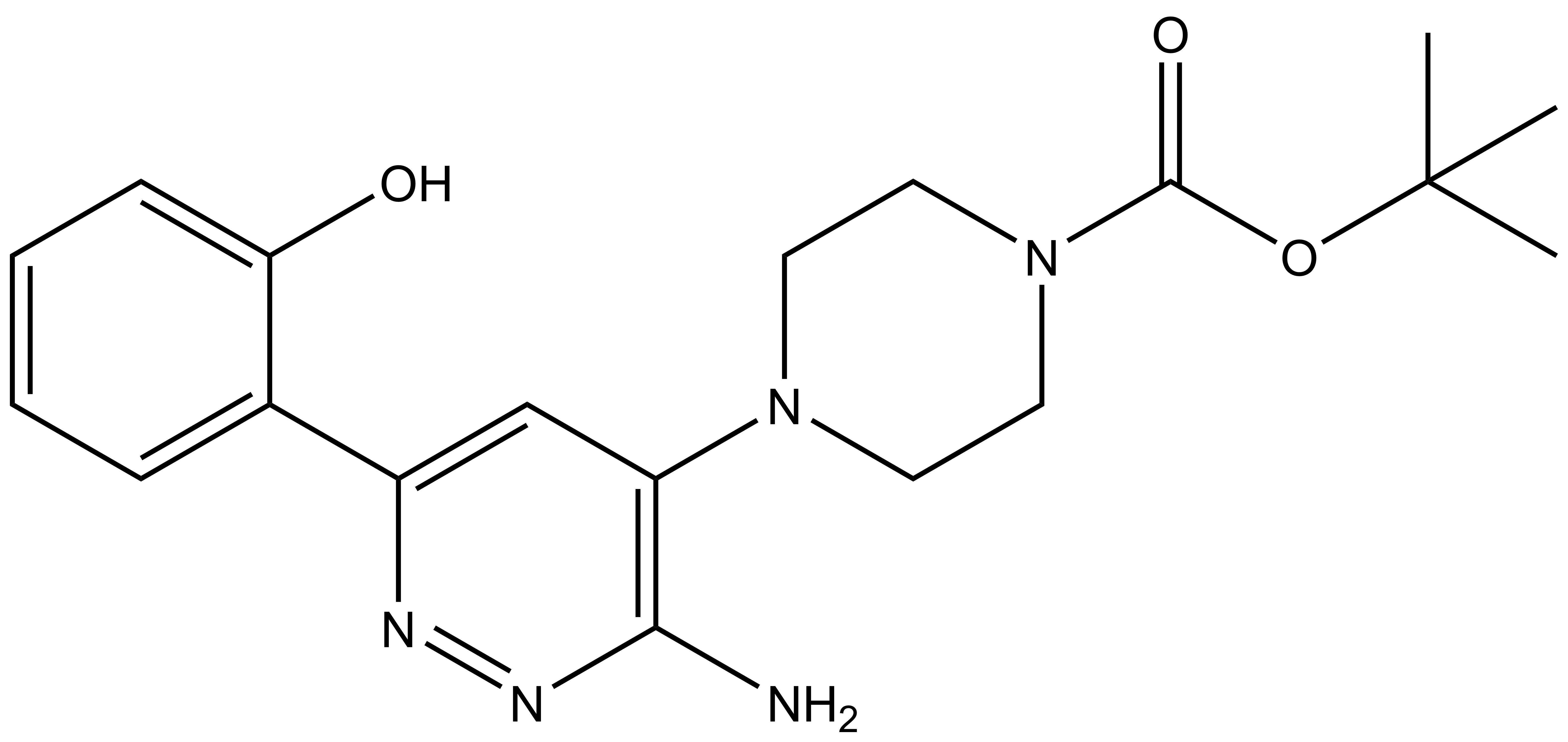
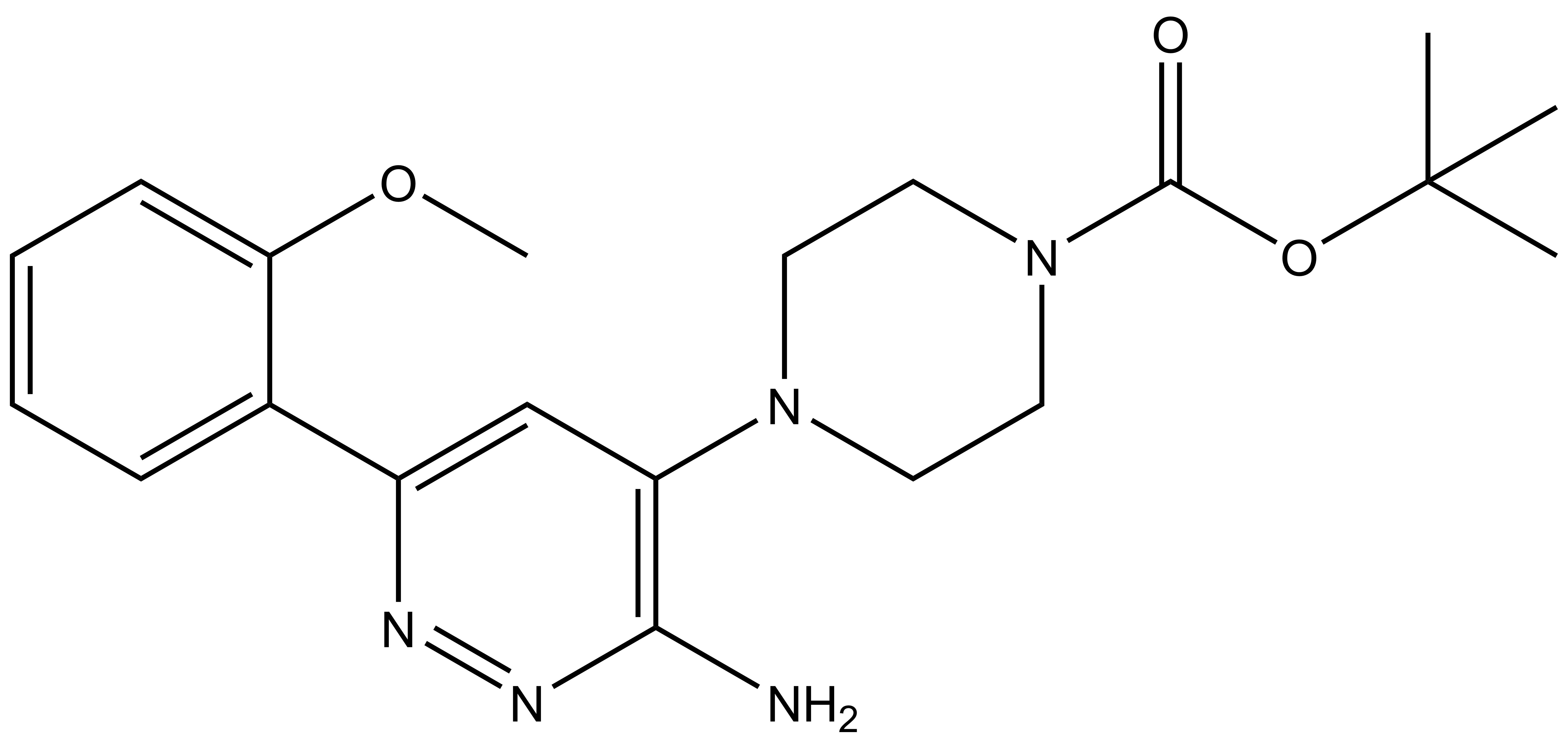
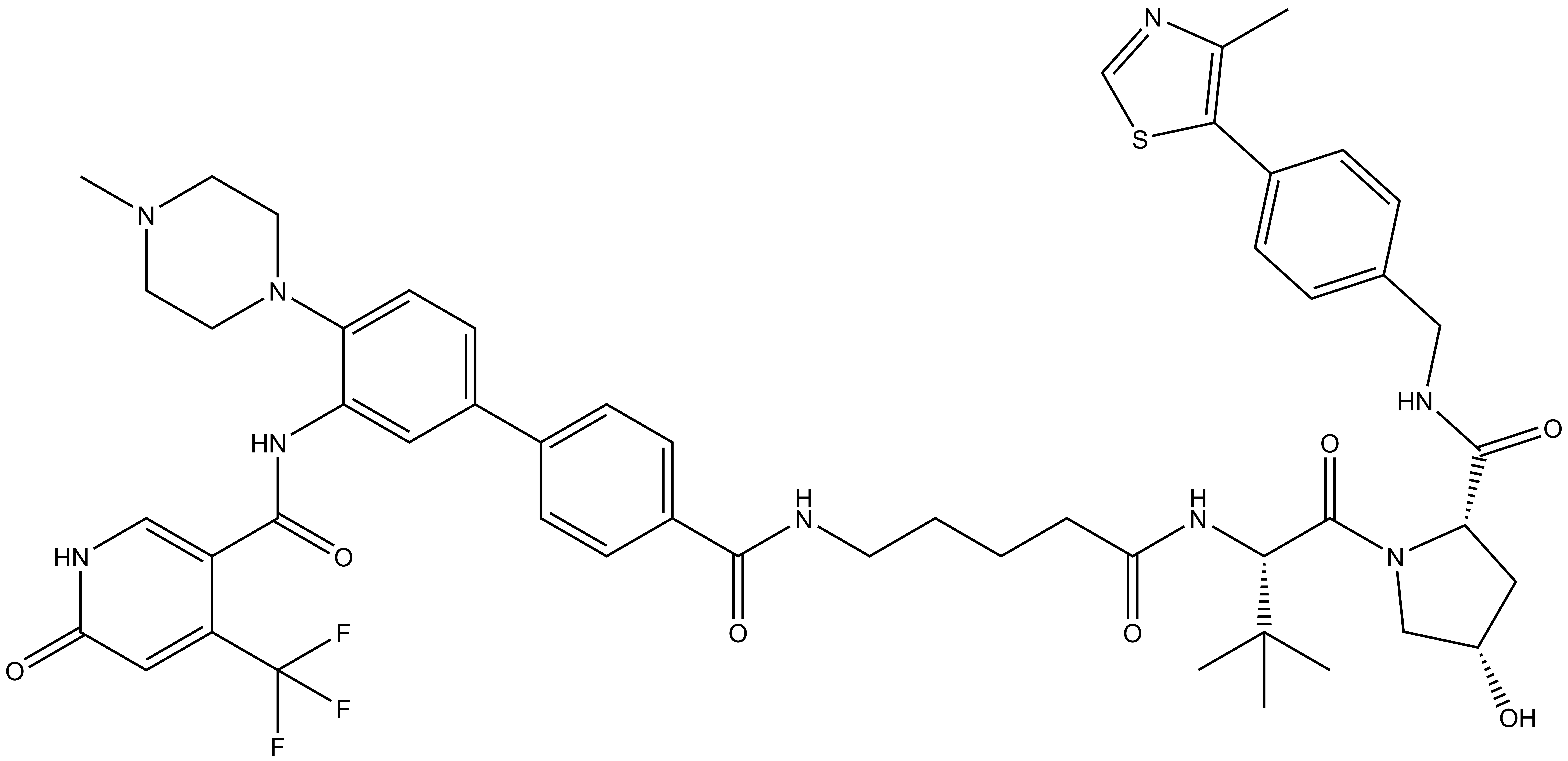
| IUPAC name | N-(4'-((5-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentyl)carbamoyl)-4-(4-methylpiperazin-1-yl)-[1,1'-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide |
| clogPo/w (SwissADME) | 5.31 |
| TPSA (SwissADME) | 237.41 Å2 |
| No. of chiral centres | 3 |
| No. of rotatable bonds | 23 |
| No. of hydrogen bond acceptors | 12 |
| No. of hydrogen bond donors | 6 |
| Storage | stable as powder at -20°C. NB making aliquots rather than freeze-thawing is recommended |
| Dissolution | soluble in DMSO at 50 mM |
| nc_VHL |
| Physical and Chemical Properties |
| Molecular weight | 1012.16 |
| Molecular formula | C52H60F3N9O7S |
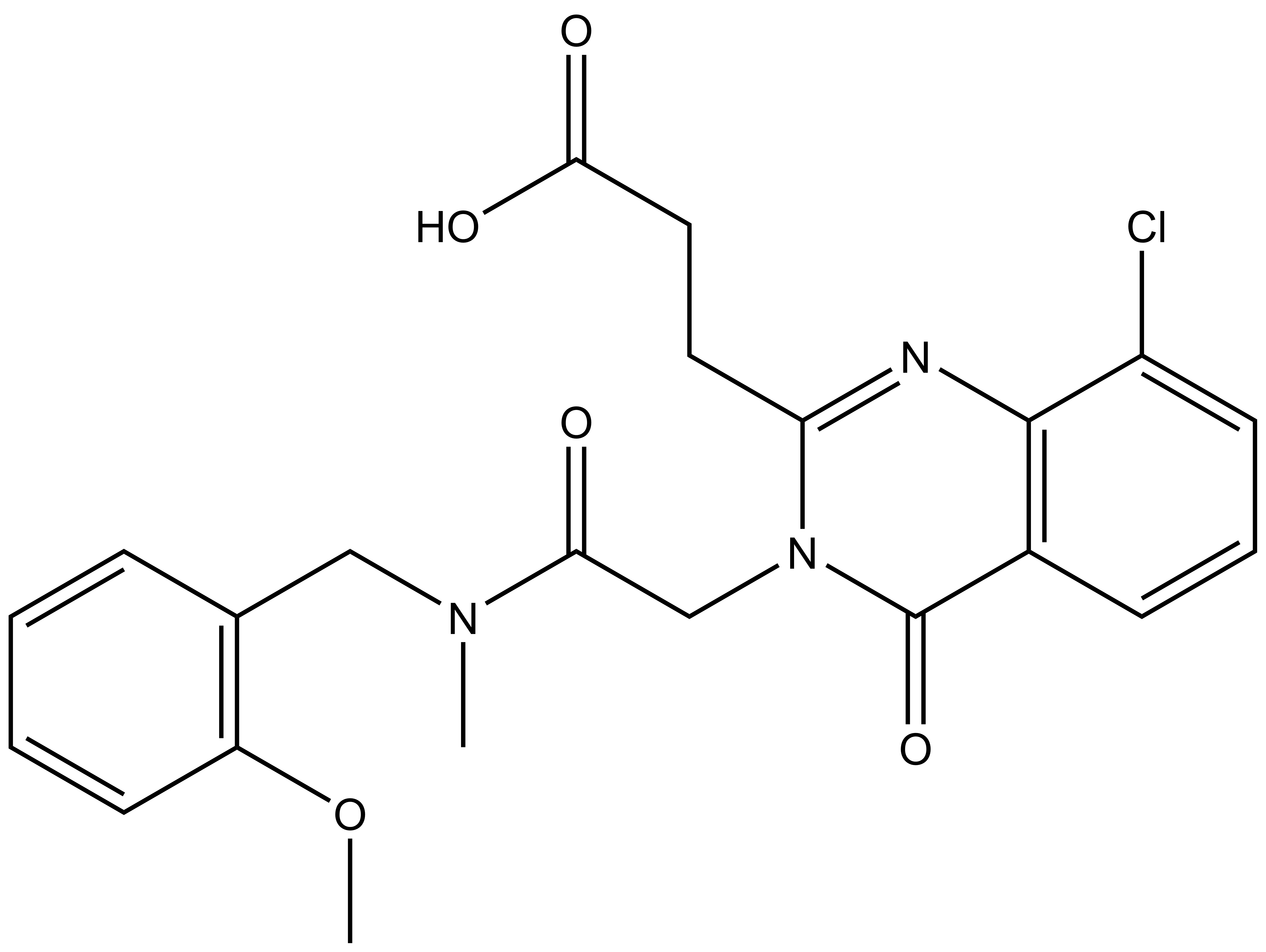
| IUPAC name | N-(4'-((5-(((S)-1-((2S,4S)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentyl)carbamoyl)-4-(4-methylpiperazin-1-yl)-[1,1'-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide |
| clogPo/w (SwissADME) | 5.25 |
| TPSA (SwissADME) | 237.41 Å2 |
| No. of chiral centres | 3 |
| No. of rotatable bonds | 23 |
| No. of hydrogen bond acceptors | 12 |
| No. of hydrogen bond donors | 6 |
| Storage | stable as powder at -20°C. NB making aliquots rather than freeze-thawing is recommended |
| Dissolution | soluble in DMSO at 50 mM |
| nc_WDR5 |
| Physical and Chemical Properties |
| Molecular weight | 999.12 |
| Molecular formula | C51H57F3N8O9S |
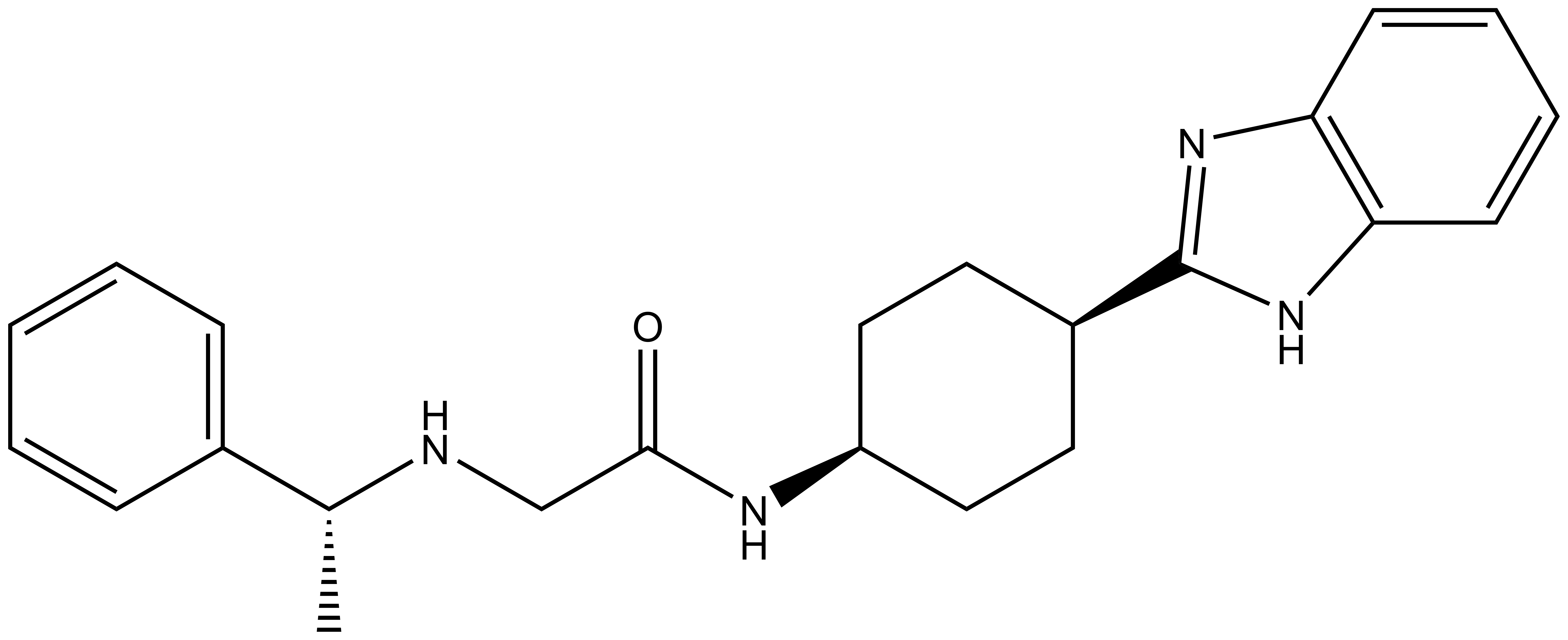
| IUPAC name | N-(4'-((5-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentyl)carbamoyl)-4-morpholino-[1,1'-biphenyl]-3-yl)-6-oxo-4-(trifluoromethyl)-1,6-dihydropyridine-3-carboxamide |
| clogPo/w (SwissADME) | 5.41 |
| TPSA (SwissADME) | 243.40 Å2 |
| No. of chiral centres | 3 |
| No. of rotatable bonds | 23 |
| No. of hydrogen bond acceptors | 12 |
| No. of hydrogen bond donors | 6 |
| Storage | stable as powder at -20°C. NB making aliquots rather than freeze-thawing is recommended |
| Dissolution | soluble in DMSO at 50 mM |
SMILES:
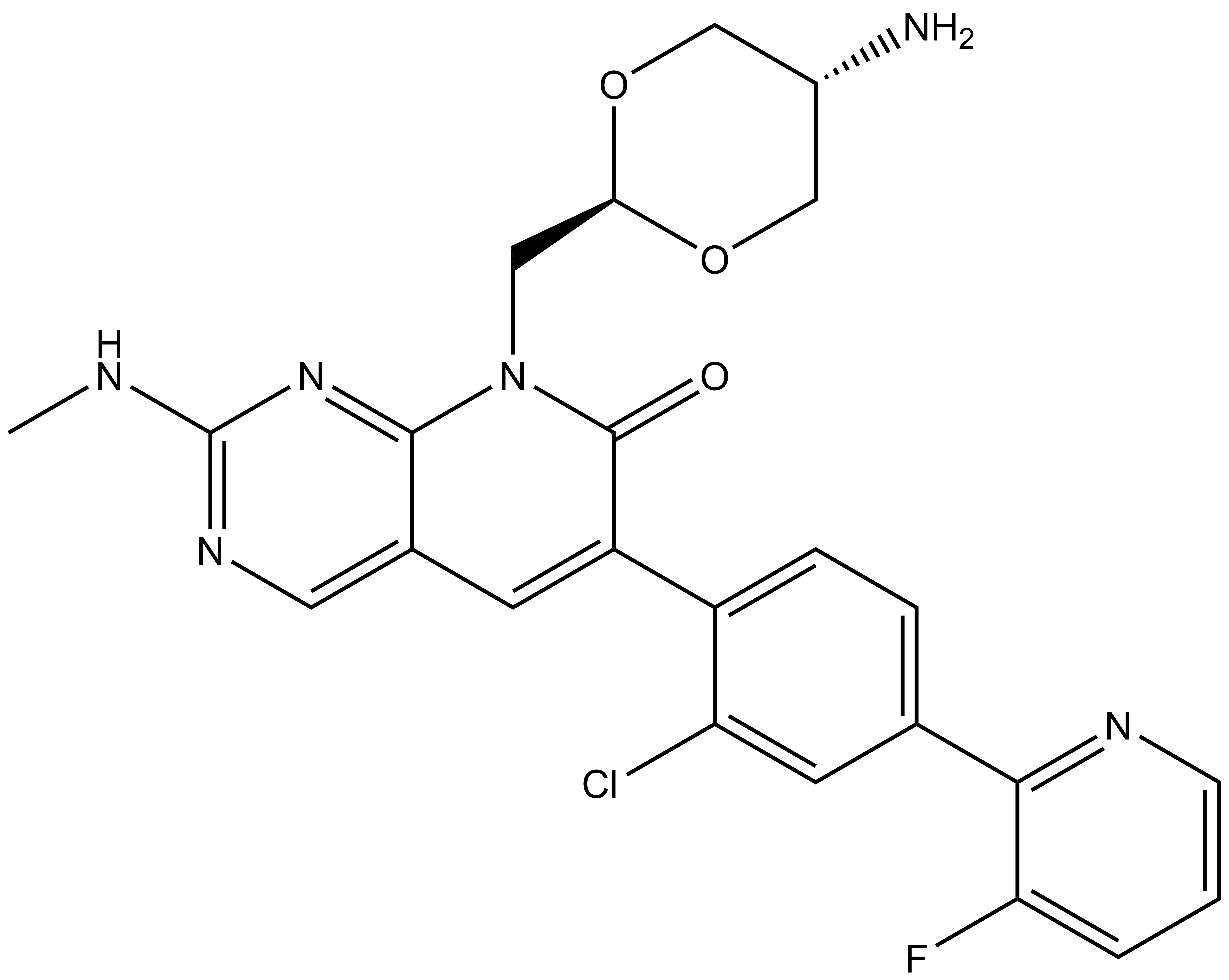
Homer: CC1=C(SC=N1)C2=CC=C(C=C2)CNC([C@@H]3C[C@H](CN3C([C@@H](NC(CCCCNC(C4=CC=C(C=C4)C5=CC=C(C(NC(C6=CNC(C=C6C(F)(F)F)=O)=O)=C5)N7CCN(CC7)C)=O)=O)C(C)(C)C)=O)O)=O
nc_VHL: CC1=C(C2=CC=C(CNC([C@@H]3C[C@H](O)CN3C([C@H](C(C)(C)C)NC(CCCCNC(C4=CC=C(C5=CC=C(N6CCN(C)CC6)C(NC(C7=CNC(C=C7C(F)(F)F)=O)=O)=C5)C=C4)=O)=O)=O)=O)C=C2)SC=N1
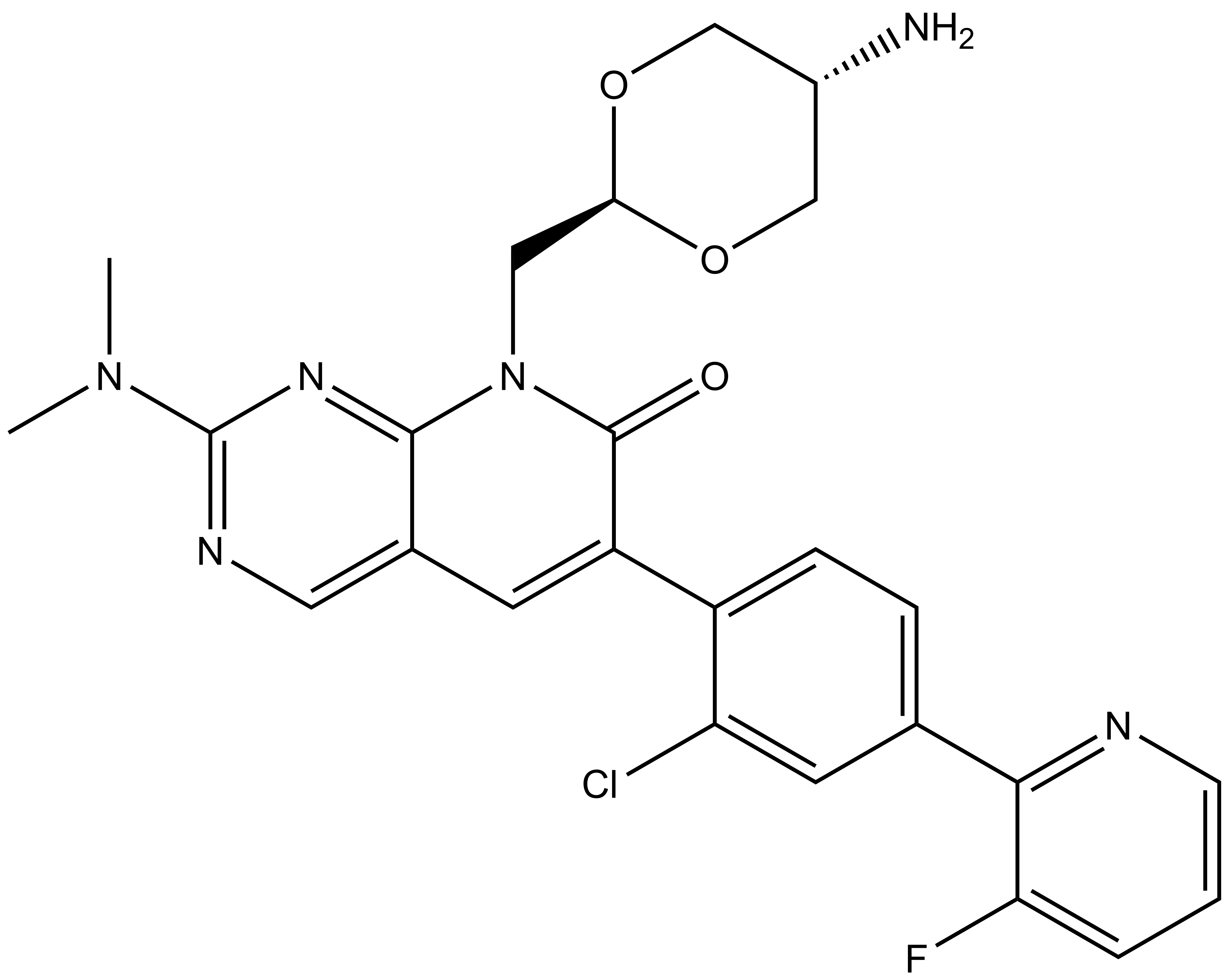
nc_WDR5: CC1=C(C2=CC=C(CNC([C@@H]3C[C@@H](O)CN3C([C@H](C(C)(C)C)NC(CCCCNC(C4=CC=C(C5=CC=C(N6CCOCC6)C(NC(C7=CNC(C=C7C(F)(F)F)=O)=O)=C5)C=C4)=O)=O)=O)=O)C=C2)SC=N1
InChI:
Homer: InChI=1S/C52H60F3N9O7S/c1-31-45(72-30-59-31)34-11-9-32(10-12-34)27-58-49(70)42-25-37(65)29-64(42)50(71)46(51(2,3)4)61-43(66)8-6-7-19-56-47(68)35-15-13-33(14-16-35)36-17-18-41(63-22-20-62(5)21-23-63)40(24-36)60-48(69)38-28-57-44(67)26-39(38)52(53,54)55/h9-18,24,26,28,30,37,42,46,65H,6-8,19-23,25,27,29H2,1-5H3,(H,56,68)(H,57,67)(H,58,70)(H,60,69)(H,61,66)/t37-,42+,46-/m1/s1
nc_VHL: InChI=1S/C52H60F3N9O7S/c1-31-45(72-30-59-31)34-11-9-32(10-12-34)27-58-49(70)42-25-37(65)29-64(42)50(71)46(51(2,3)4)61-43(66)8-6-7-19-56-47(68)35-15-13-33(14-16-35)36-17-18-41(63-22-20-62(5)21-23-63)40(24-36)60-48(69)38-28-57-44(67)26-39(38)52(53,54)55/h9-18,24,26,28,30,37,42,46,65H,6-8,19-23,25,27,29H2,1-5H3,(H,56,68)(H,57,67)(H,58,70)(H,60,69)(H,61,66)/t37-,42-,46+/m0/s1
nc_WDR5: InChI=1S/C51H57F3N8O8S/c1-30-44(71-29-58-30)33-10-8-31(9-11-33)26-57-48(68)41-24-36(63)28-62(41)49(69)45(50(2,3)4)60-42(64)7-5-6-18-55-46(66)34-14-12-32(13-15-34)35-16-17-40(61-19-21-70-22-20-61)39(23-35)59-47(67)37-27-56-43(65)25-38(37)51(52,53)54/h8-17,23,25,27,29,36,41,45,63H,5-7,18-22,24,26,28H2,1-4H3,(H,55,66)(H,56,65)(H,57,68)(H,59,67)(H,60,64)/t36-,41+,45-/m1/s1
InChIKey:
Homer: OFNZESNEBSQKSE-BQGOKDIQSA-N
nc_VHL: OFNZESNEBSQKSE-CCVFZADKSA-N
nc_WDR5: BFXBMIZHEIFXOC-LTJJNQLXSA-N