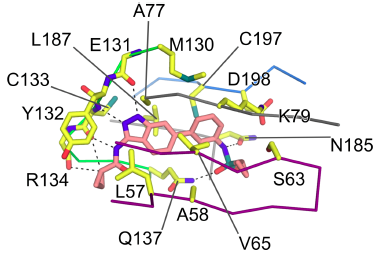
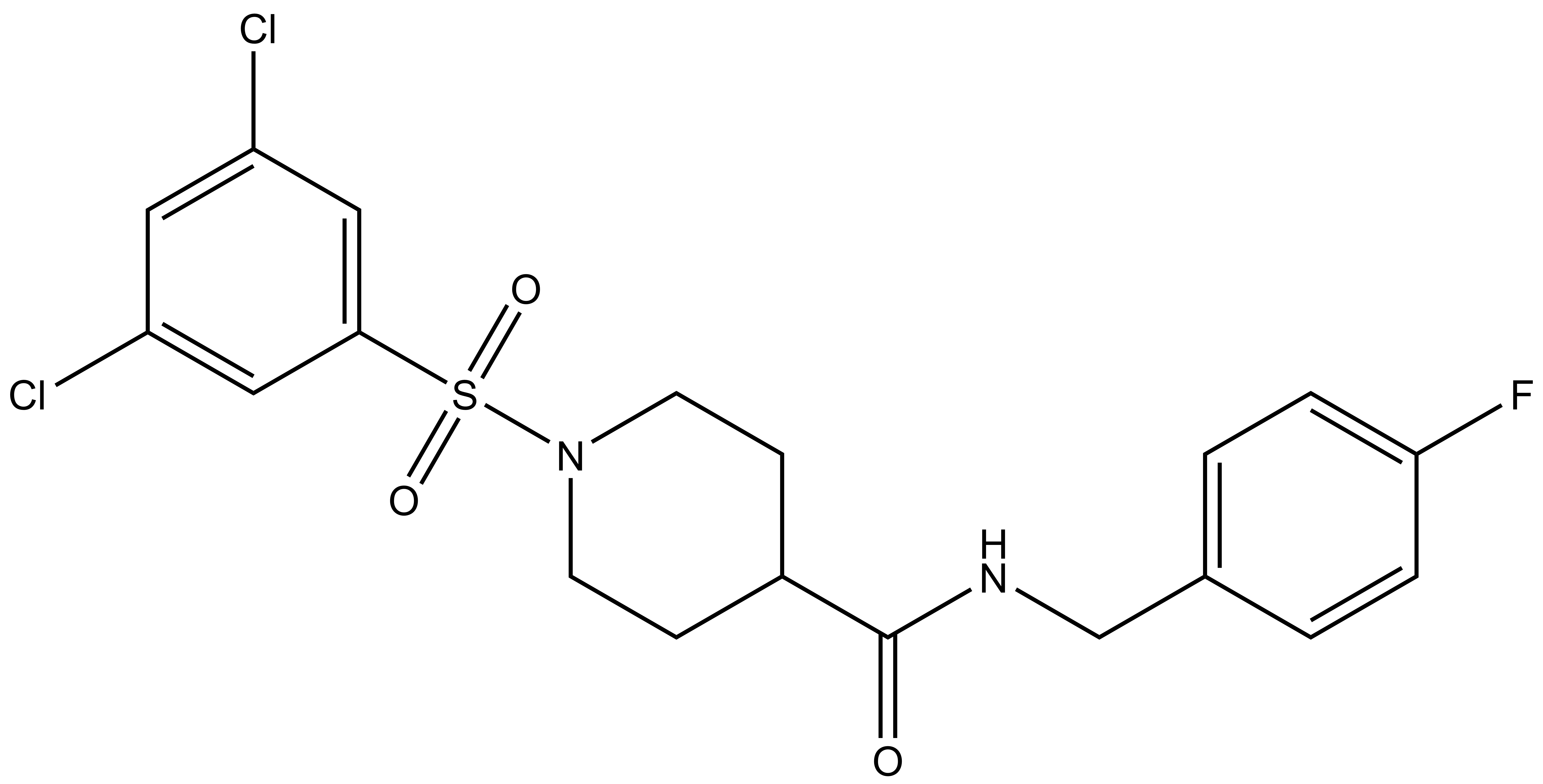
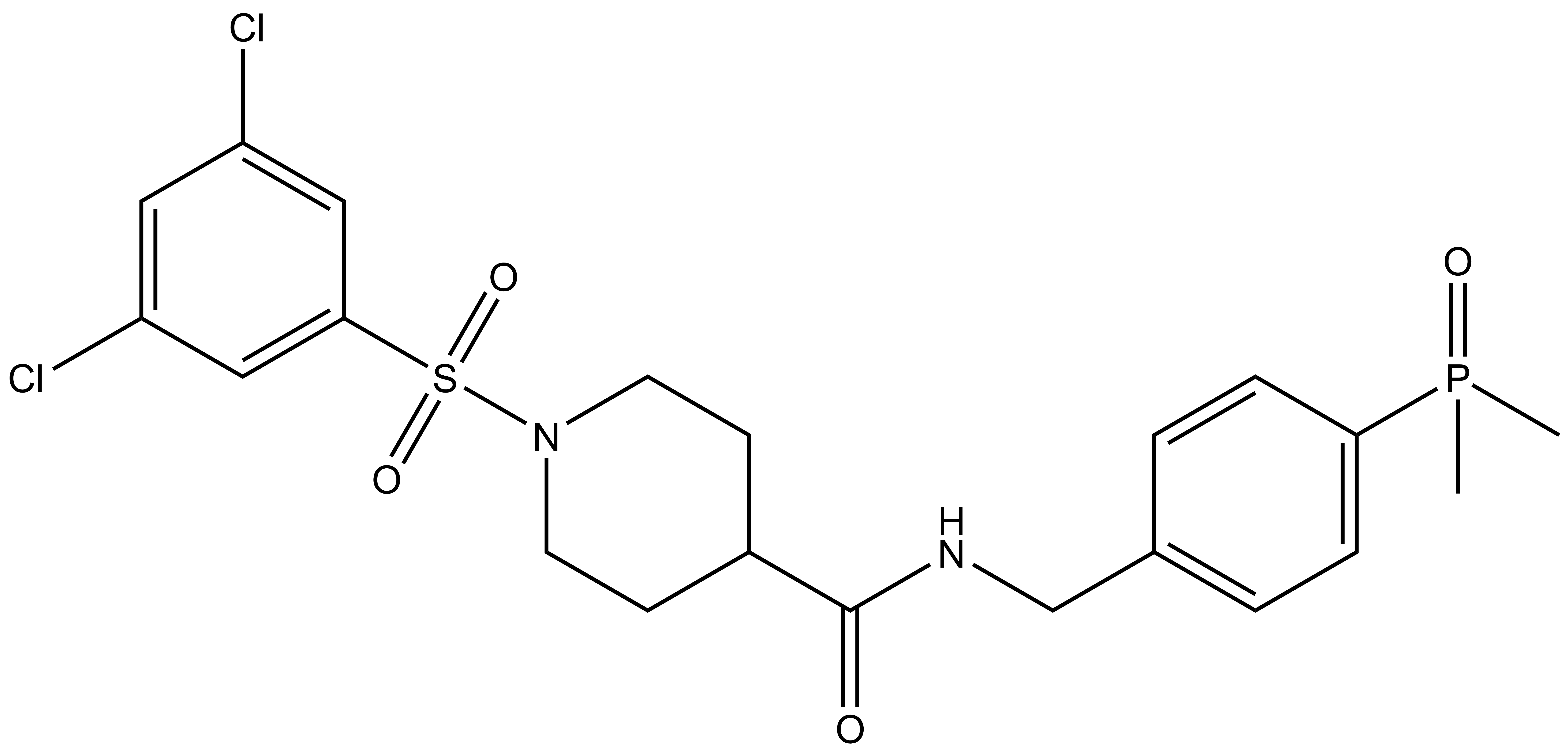
| Probe | | Negative control |
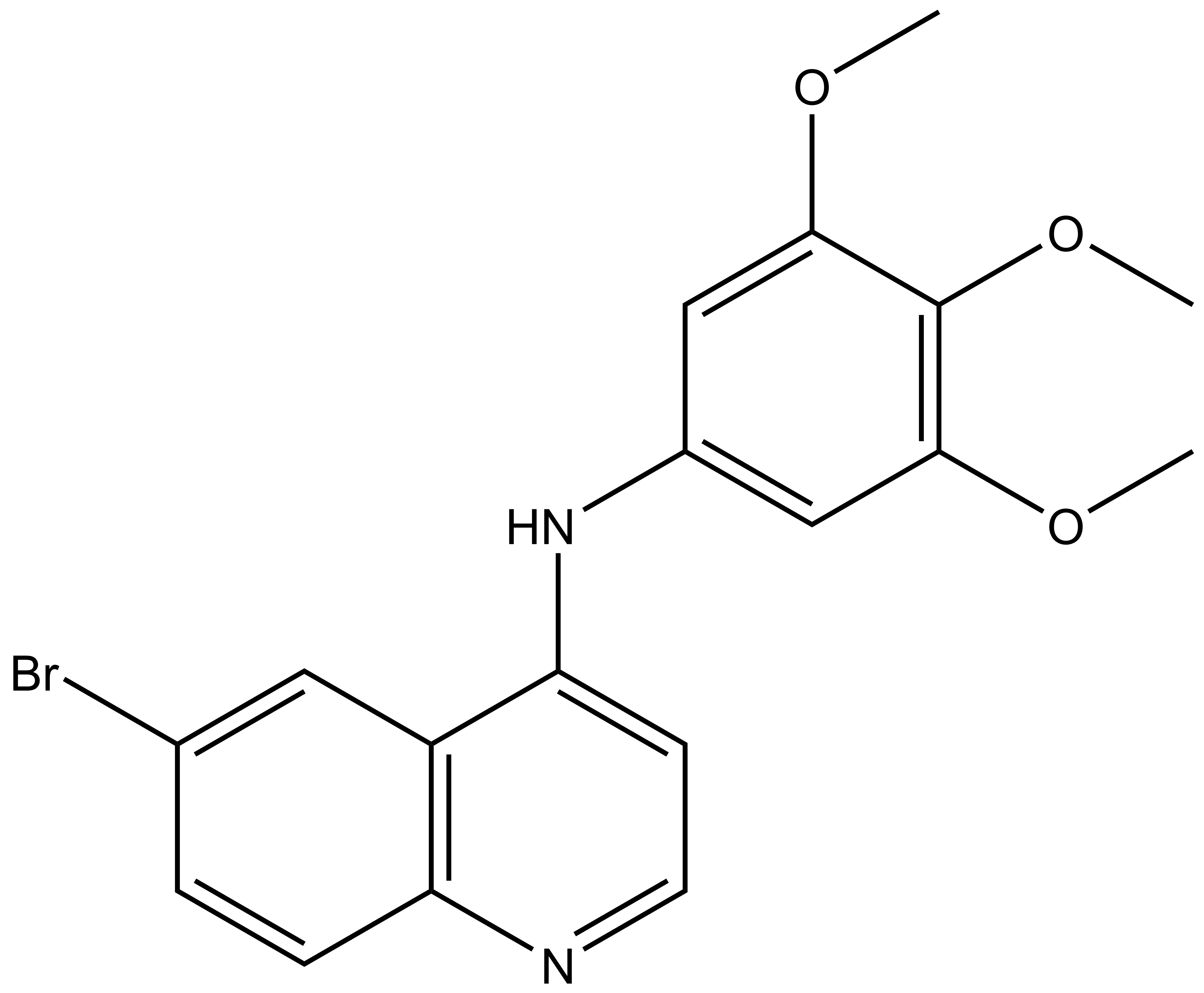 | | 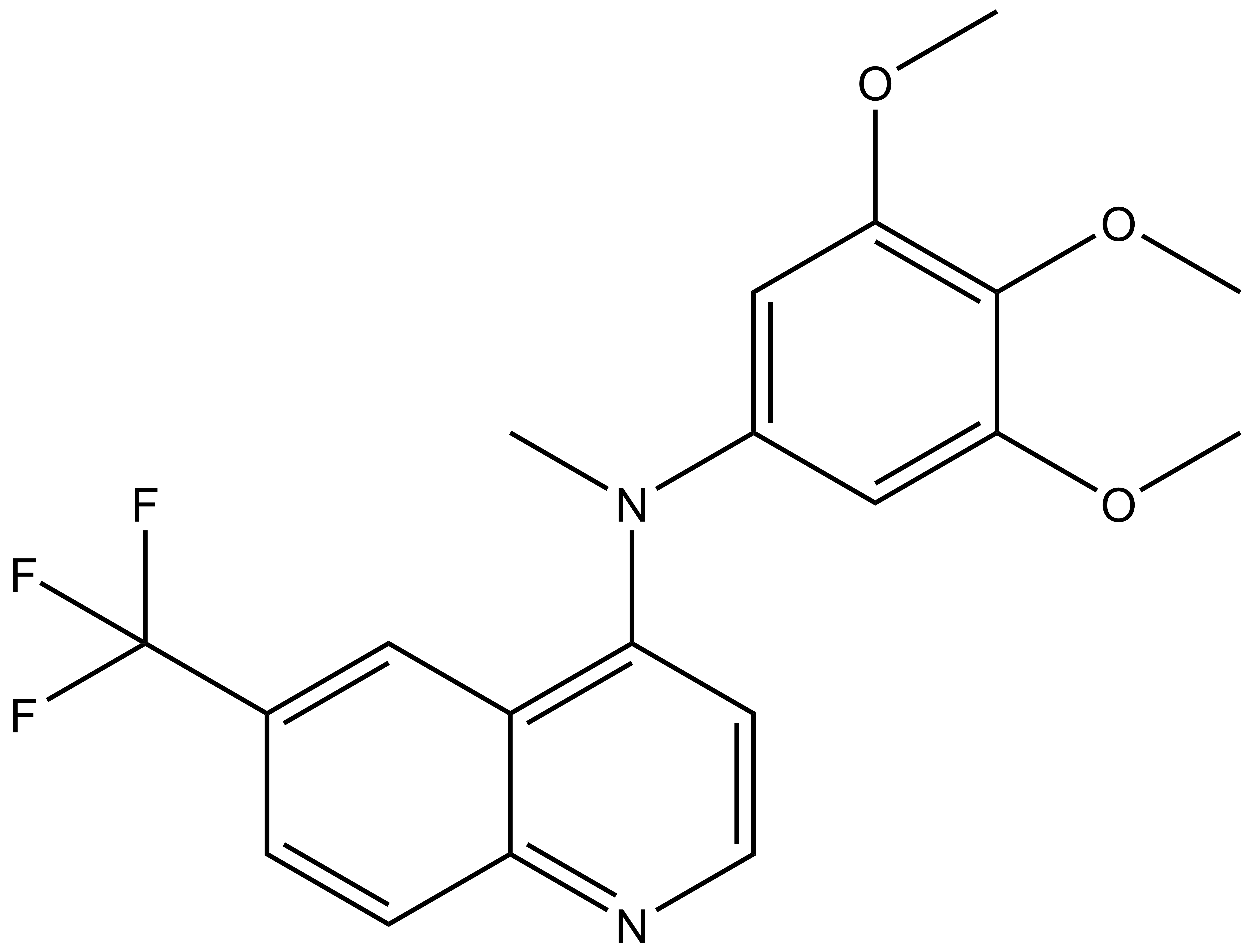 |
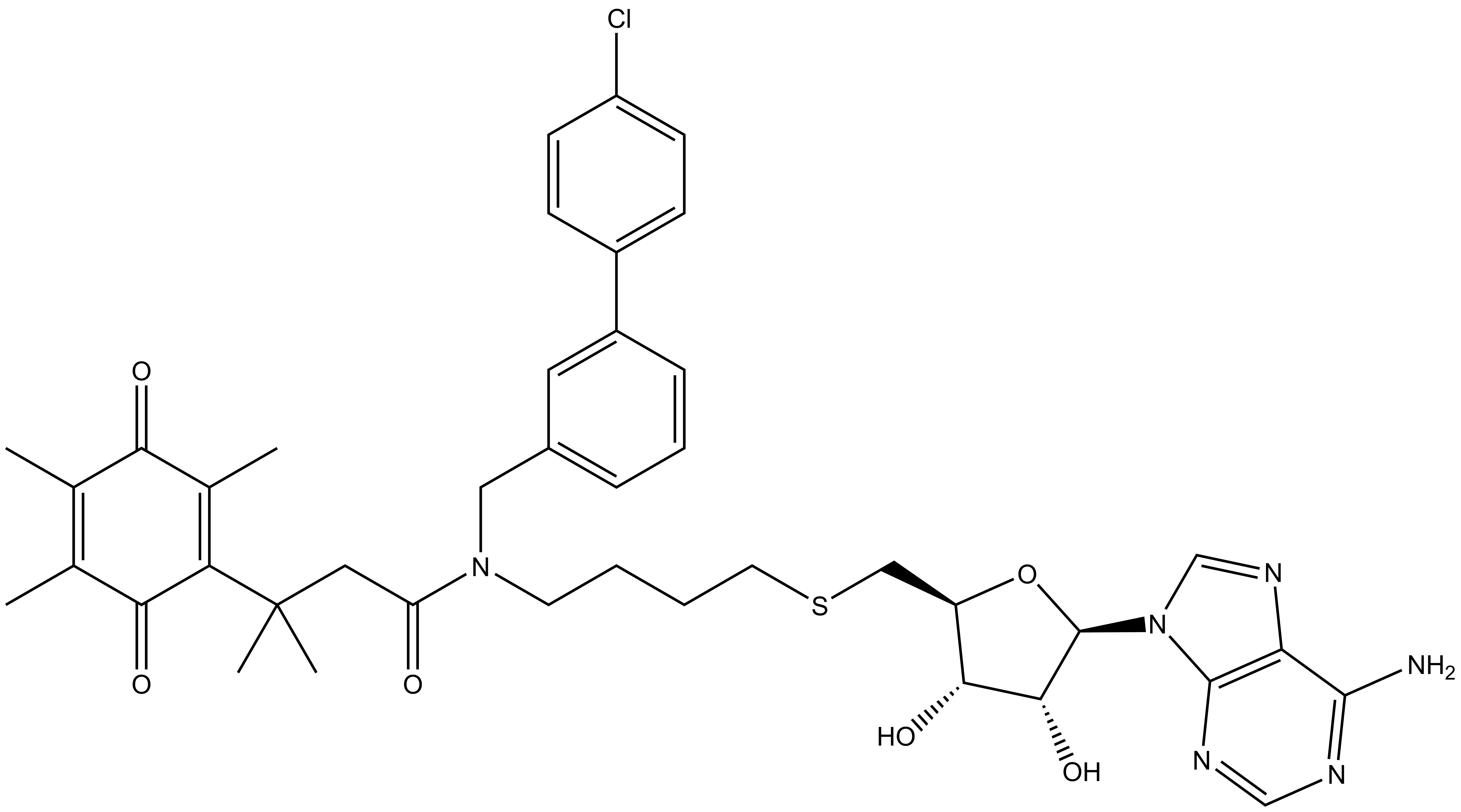
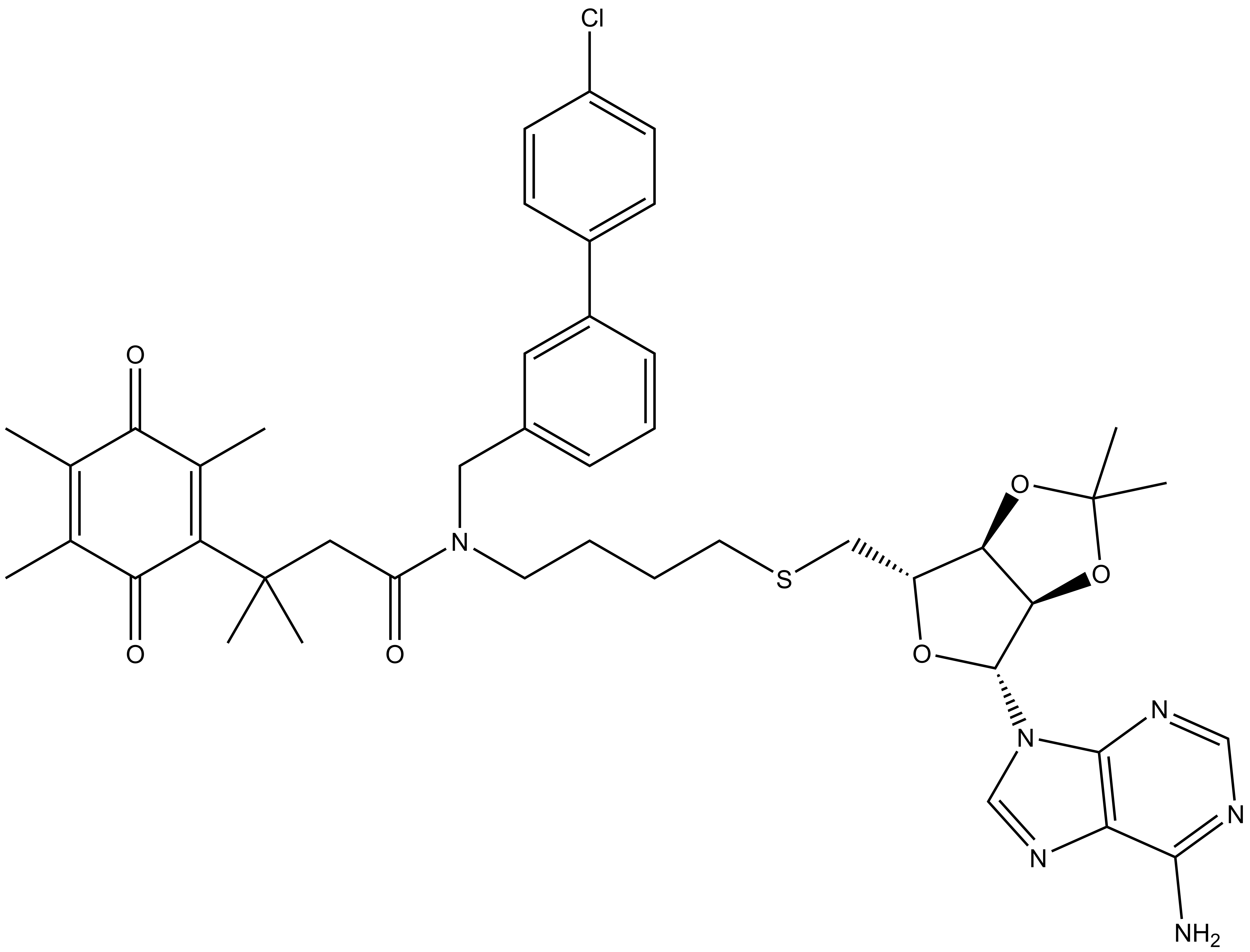
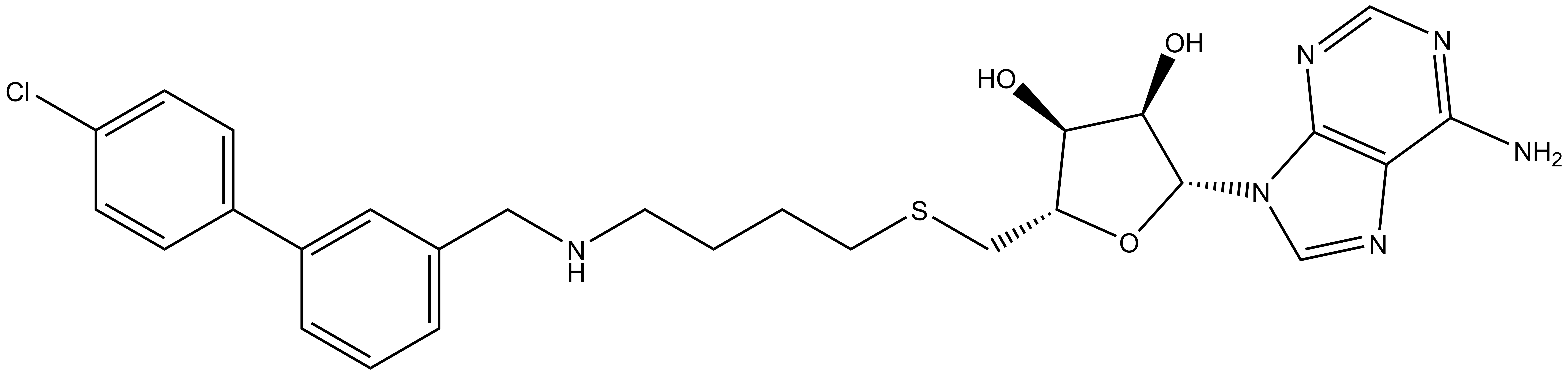
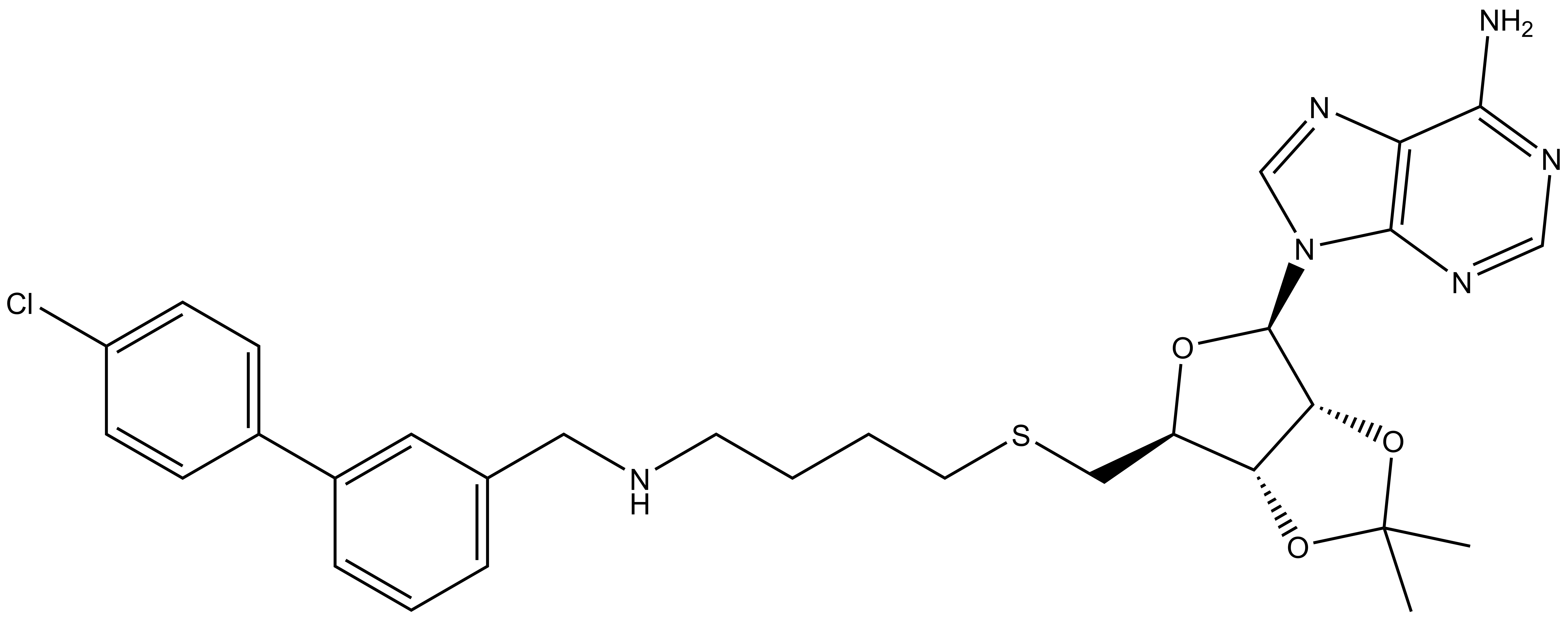
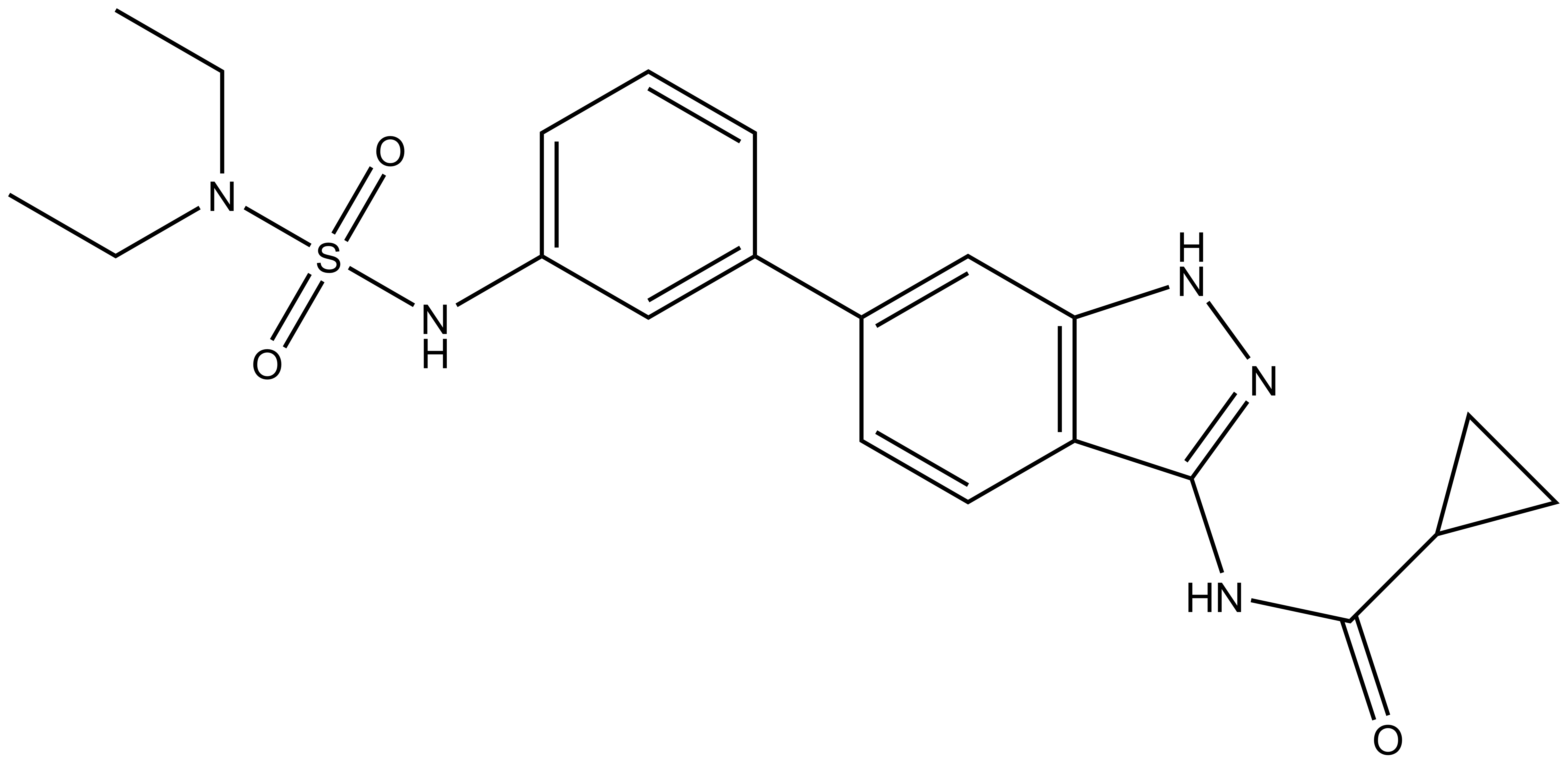
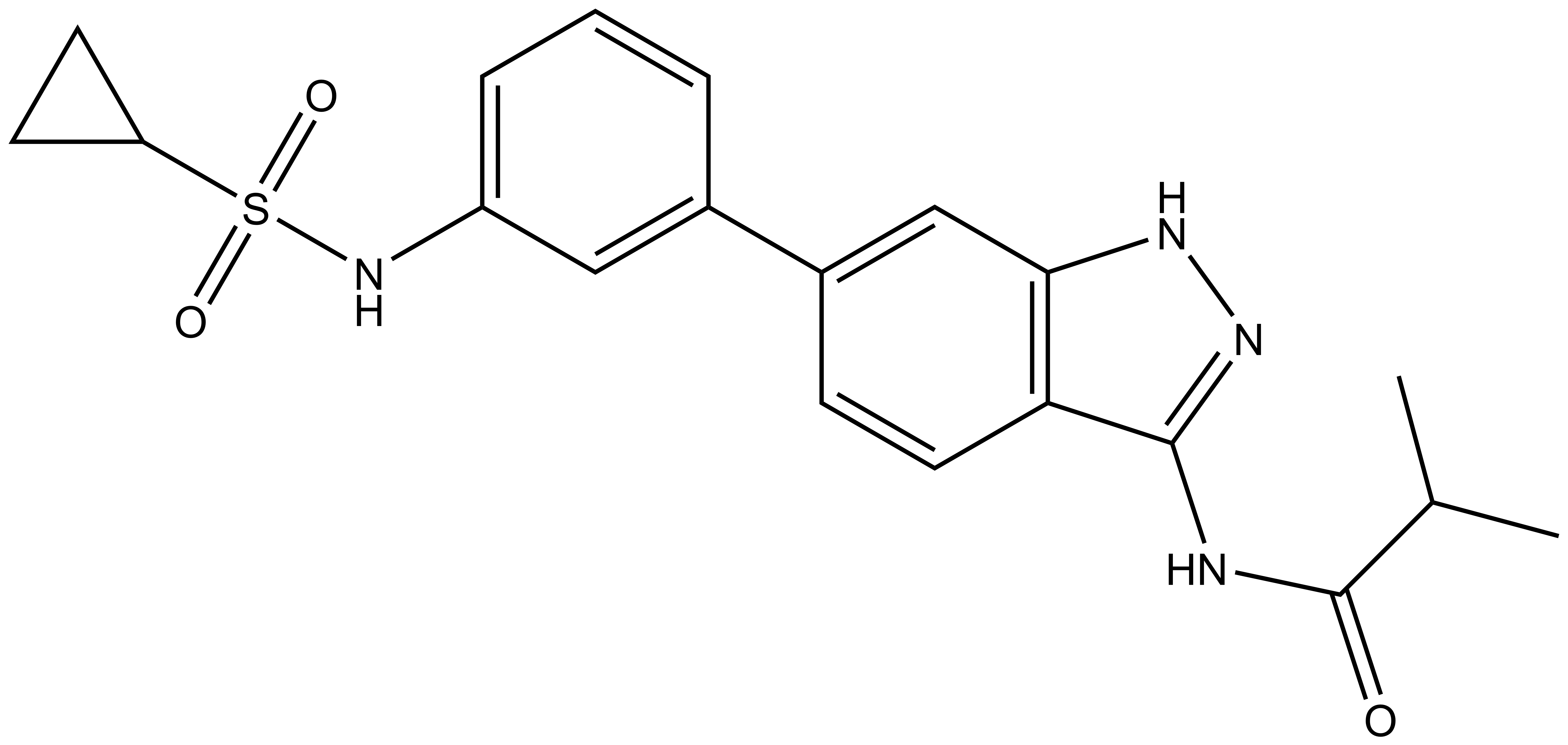
SGC-GAK-1 | | SGC-GAK-1N |
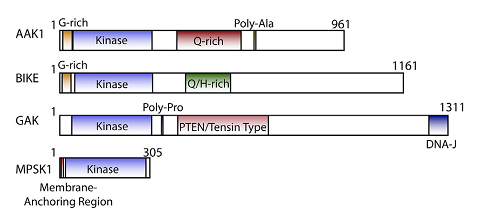
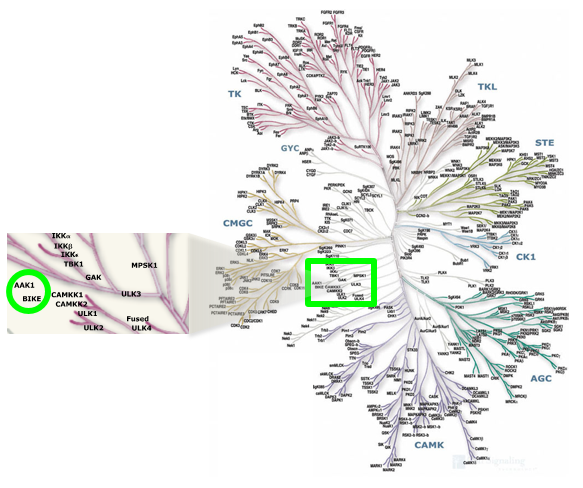
Cyclin G associated kinase (GAK) is a 160 kDa serine/threonine kinase originally identified and so named as a direct association partner with cyclin G.1 GAK is a member of the numb-associated kinase (NAK) family, which includes AAK1 (adaptor protein 2-associated kinase), STK16/MPSK1 (serine/threonine kinase 16/myristoylated and palmitoylated serine/threonine kinase 1), and BMP2K/BIKE (BMP-2 inducible kinase).2 In addition to its kinase domain, the C-terminus of GAK protein bears high homology to a domain found in auxilin and tensin.3 GAK has ubiquitous tissue expression and within the cell localizes to the Golgi complex, cytoplasm, and nucleus.
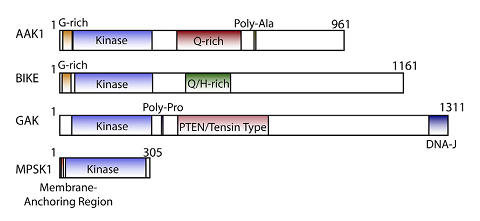
NAK family domain structures
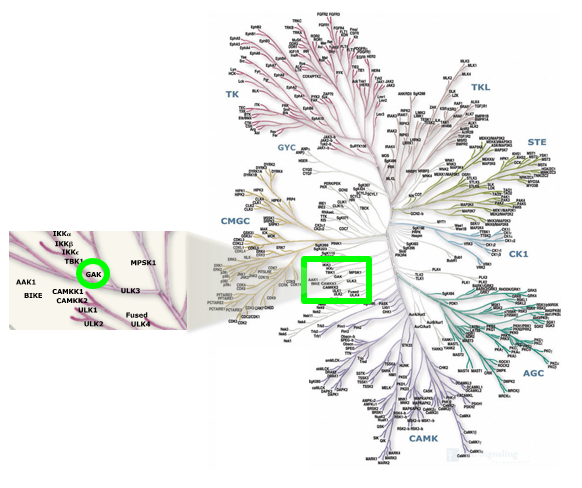
Location of GAK on kinome tree
Multiple biological roles for GAK and disease associations have been made despite it being a relatively understudied kinase. GAK is involved in membrane trafficking and sorting of proteins, including as an essential cofactor for HSC70-dependent uncoating of clathrin coated vesicles in the cytoplasm.6-7 GAK is required for maintenance of centrosome maturation and progression through mitosis.8 GAK is over-expressed in osteosarcoma cell lines and tissues where it contributes to proliferation and survival.9 Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms in the GAK gene associated with susceptibility to Parkinson’s disease.10 Emerging evidence suggests that GAK may be a therapeutic target in prostate cancer (PCa): GAK expression levels increase upon prolonged androgen treatment and during the progression of cells to hormone independence, and analysis of patient-derived tissue samples demonstrated that GAK expression levels positively correlated with the Gleason score, a quantitative scale of PCa aggressiveness.11 GAK interacts directly with the androgen receptor (AR) and potentiates its transcriptional activity.12 Further elucidation of the role of GAK in these and other biological processes would be facilitated by access to a potent and selective inhibitor of the kinase.
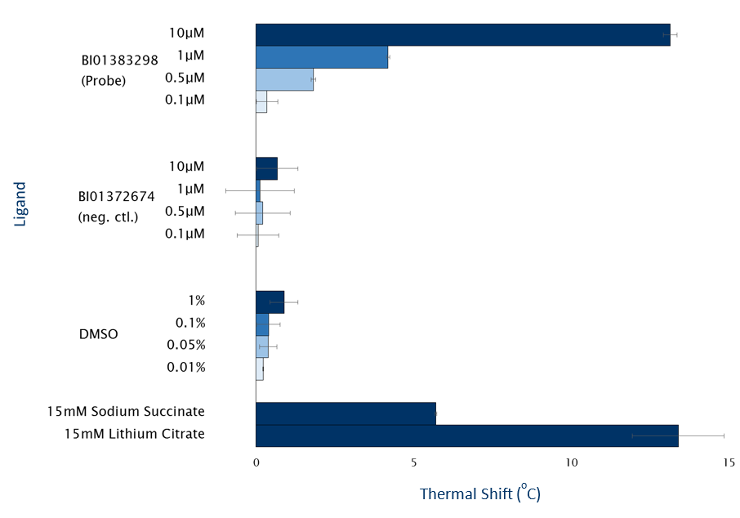
SGC-GAK-1 is a chemical probe for GAK that potently targets the ATP-binding site (KD = 1.9 nM). Regarding kinase selectivity, no kinases were observed to bind SGC-GAK-1 within 30-fold of the KD of GAK in a KINOMEscan assay (DiscoverX) at 1 μM followed by KD determinations. In a live cell NanoBRET assay (Promega) SGC-GAK-1 had an IC50 of 110 nM against ectopically expressed full-length GAK-Nluc fusion.
A chemically related negative control compound, SGC-GAK-1N, is provided.
Despite weakly binding RIPK2 in weakly binding RIPK2 in vitro (DiscoverX RIPK2 KD = 110 nM; 58-fold of GAK KD), SGC-GAK-1 potently engaged RIPK2 in a live cell NanoBRET assay (RIPK2 IC50 = 360 nM); accordingly, we have identified a control compound that is a highly cell-potent RIPK2 ligand, HY-19764, (IC50 = 2.2 nM) that lacks GAK affinity (IC50 > 10 µM).